

Oxidative Stress in Placenta: Health and Diseases
- PMID: 26693479
- PMCID: PMC4676991
- DOI: 10.1155/2015/293271
Oxidative Stress in Placenta: Health and Diseases
Abstract
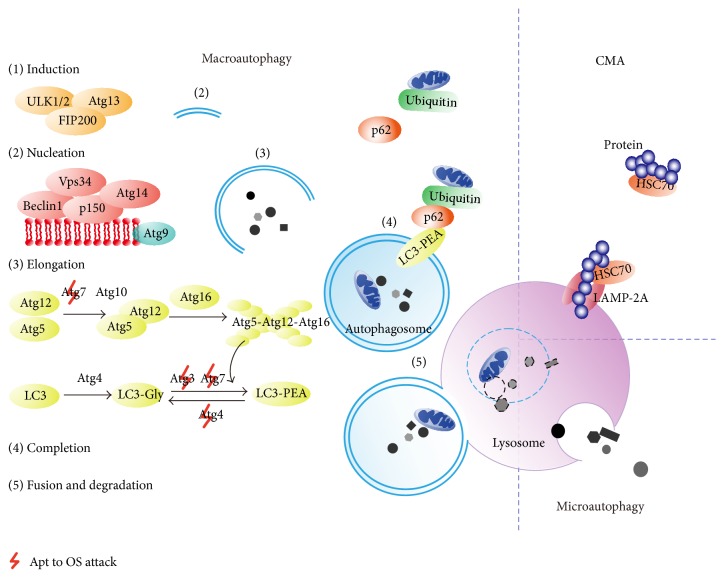
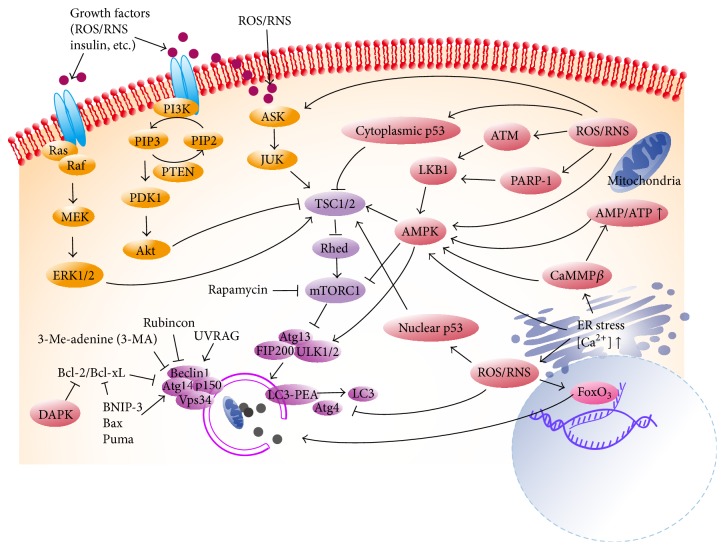
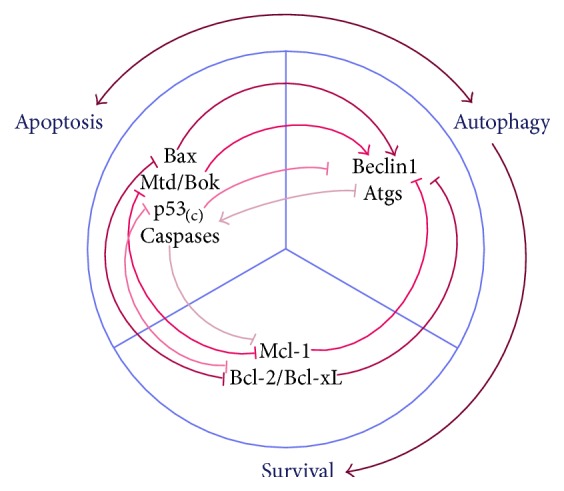
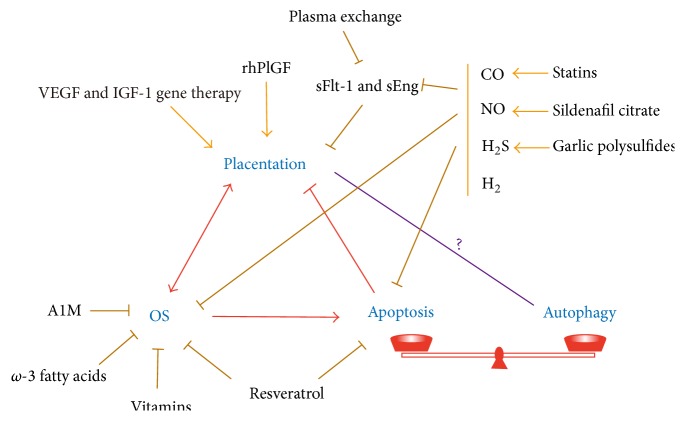
During pregnancy, development of the placenta is interrelated with the oxygen concentration. Embryo development takes place in a low oxygen environment until the beginning of the second trimester when large amounts of oxygen are conveyed to meet the growth requirements. High metabolism and oxidative stress are common in the placenta. Reactive oxidative species sometimes harm placental development, but they are also reported to regulate gene transcription and downstream activities such as trophoblast proliferation, invasion, and angiogenesis. Autophagy and apoptosis are two crucial, interconnected processes in the placenta that are often influenced by oxidative stress. The proper interactions between them play an important role in placental homeostasis. However, an imbalance between the protective and destructive mechanisms of autophagy and apoptosis seems to be linked with pregnancy-related disorders such as miscarriage, preeclampsia, and intrauterine growth restriction. Thus, potential therapies to hold oxidative stress in leash, promote placentation, and avoid unwanted apoptosis are discussed.
Figures
References
-
- Ďuračková Z. Some current insights into oxidative stress. Physiological Research. 2010;59(4):459–469. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
