

Sporadic and Familial Congenital Cataracts: Mutational Spectrum and New Diagnoses Using Next-Generation Sequencing
- PMID: 26694549
- PMCID: PMC4787201
- DOI: 10.1002/humu.22948
Sporadic and Familial Congenital Cataracts: Mutational Spectrum and New Diagnoses Using Next-Generation Sequencing
Abstract
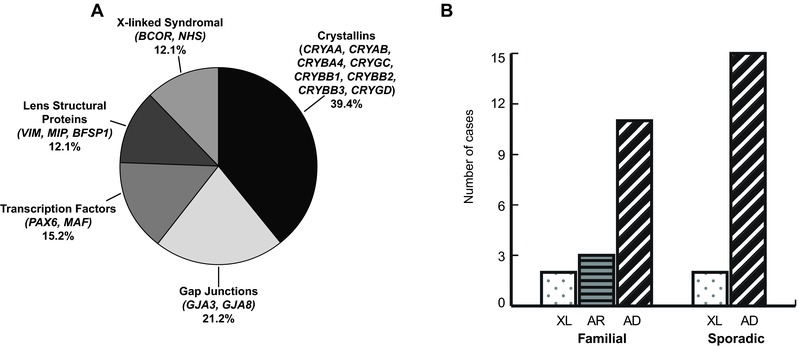
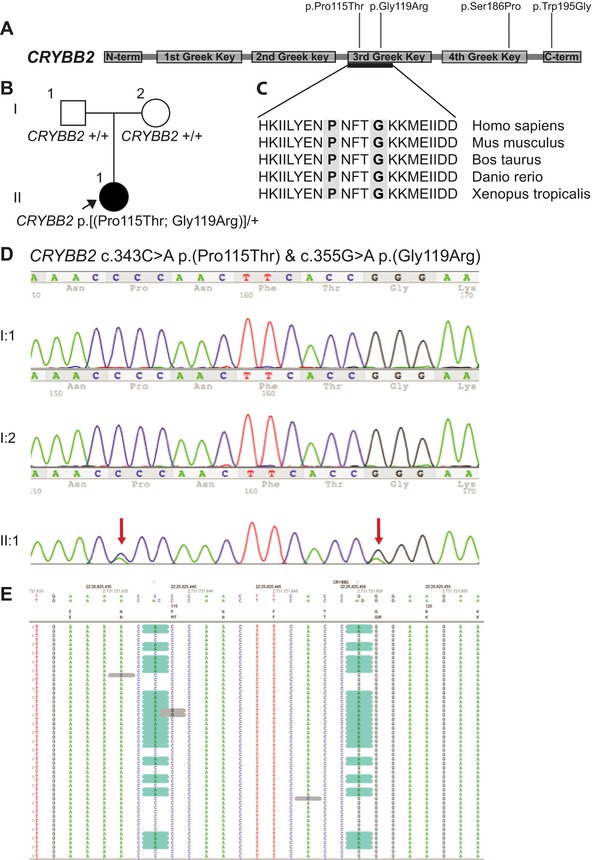
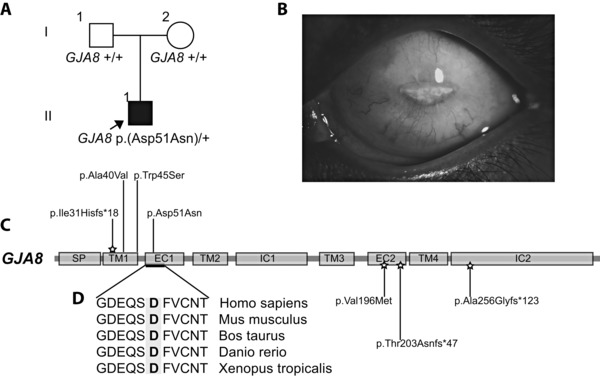
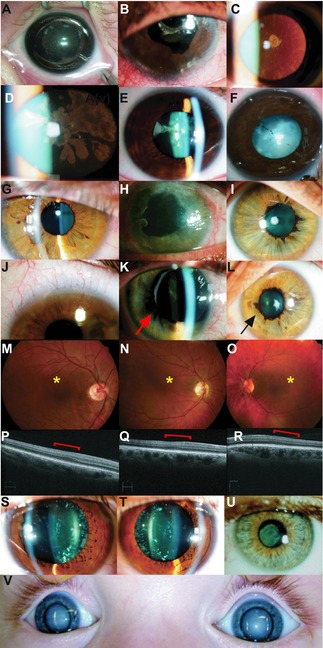
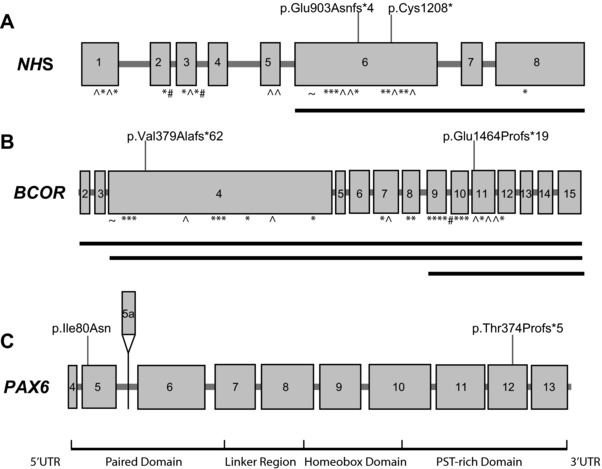
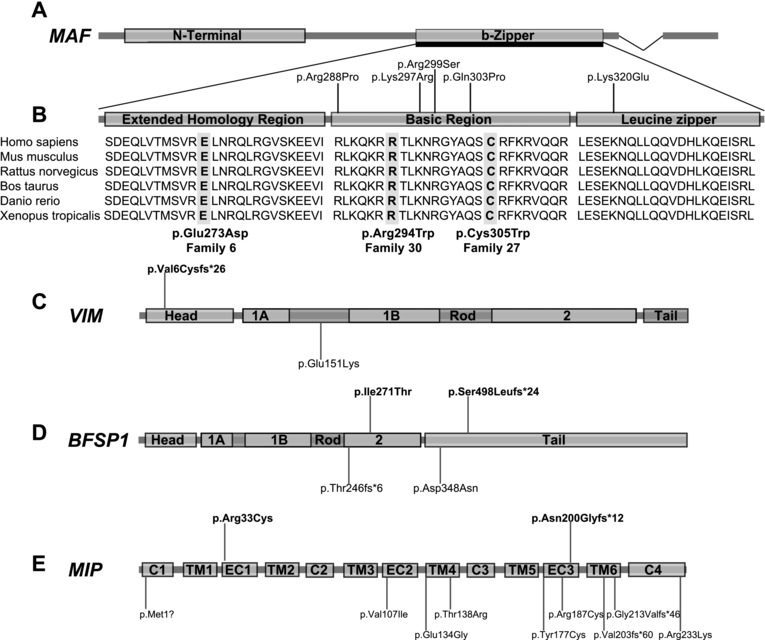
Congenital cataracts are a significant cause of lifelong visual loss. They may be isolated or associated with microcornea, microphthalmia, anterior segment dysgenesis (ASD) and glaucoma, and there can be syndromic associations. Genetic diagnosis is challenging due to marked genetic heterogeneity. In this study, next-generation sequencing (NGS) of 32 cataract-associated genes was undertaken in 46 apparently nonsyndromic congenital cataract probands, around half sporadic and half familial cases. We identified pathogenic variants in 70% of cases, and over 68% of these were novel. In almost two-thirds (20/33) of these cases, this resulted in new information about the diagnosis and/or inheritance pattern. This included identification of: new syndromic diagnoses due to NHS or BCOR mutations; complex ocular phenotypes due to PAX6 mutations; de novo autosomal-dominant or X-linked mutations in sporadic cases; and mutations in two separate cataract genes in one family. Variants were found in the crystallin and gap junction genes, including the first report of severe microphthalmia and sclerocornea associated with a novel GJA8 mutation. Mutations were also found in rarely reported genes including MAF, VIM, MIP, and BFSP1. Targeted NGS in presumed nonsyndromic congenital cataract patients provided significant diagnostic information in both familial and sporadic cases.
Keywords: congenital cataract; eye; microcornea; microphthalmia; next-generation sequencing.
© 2015 The Authors. **Human Mutation published by Wiley Periodicals, Inc.
Figures
References
-
- Bateman JB, von‐Bischhoffshaunsen FR, Richter L, Flodman P, Burch D, Spence MA. 2007. Gene conversion mutation in crystallin, beta‐B2 (CRYBB2) in a Chilean family with autosomal dominant cataract. Ophthalmology 114:425–432. - PubMed
-
- Bennett TM, Mackay DS, Knopf HL, Shiels A. 2004. A novel missense mutation in the gene for gap‐junction protein alpha3 (GJA3) associated with autosomal dominant "nuclear punctate" cataracts linked to chromosome 13q. Mol Vis 10:376–382. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
