

PrimPol-deficient cells exhibit a pronounced G2 checkpoint response following UV damage
- PMID: 26694751
- PMCID: PMC4889237
- DOI: 10.1080/15384101.2015.1128597
PrimPol-deficient cells exhibit a pronounced G2 checkpoint response following UV damage
Abstract
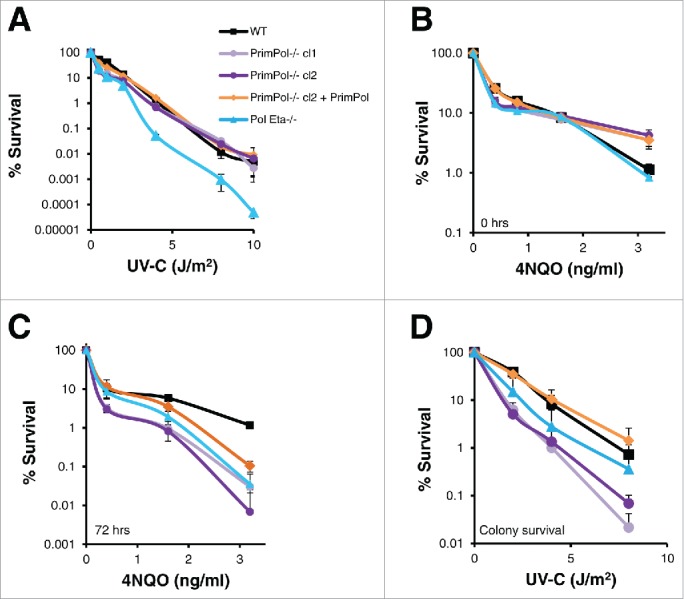
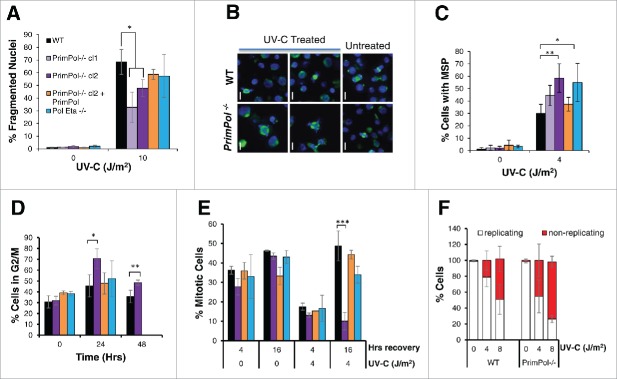
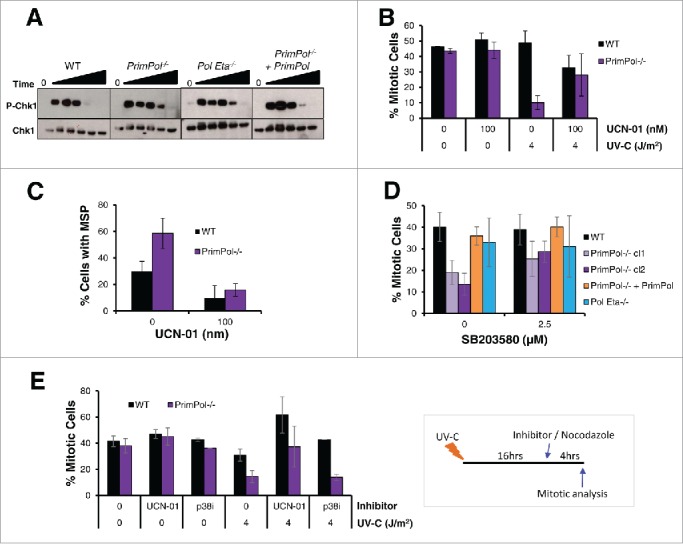
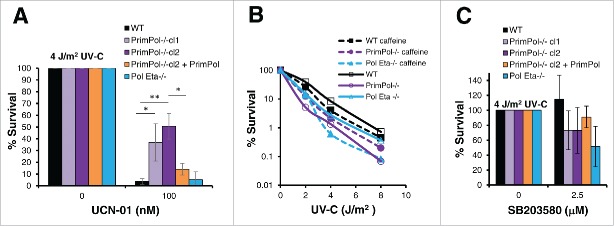
PrimPol is a recently identified member of the archaeo-eukaryote primase (AEP) family of primase-polymerases. It has been shown that this mitochondrial and nuclear localized enzyme plays roles in the maintenance of both unperturbed replication fork progression and in the bypass of lesions after DNA damage. Here, we utilized an avian (DT40) knockout cell line to further study the consequences of loss of PrimPol (PrimPol(-/-)) on the downstream maintenance of cells after UV damage. We report that PrimPol(-/-) cells are more sensitive to UV-C irradiation in colony survival assays than Pol η-deficient cells. Although this increased UV sensitivity is not evident in cell viability assays, we show that this discrepancy is due to an enhanced checkpoint arrest after UV-C damage in the absence of PrimPol. PrimPol(-/-) arrested cells become stalled in G2, where they are protected from UV-induced cell death. Despite lacking an enzyme required for the bypass and maintenance of replication fork progression in the presence of UV damage, we show that PrimPol(-/-) cells actually have an advantage in the presence of a Chk1 inhibitor due to their slow progression through S-phase.
Keywords: Chk1; DT40; PrimPol; TLS; UV; cell cycle; checkpoint; polymerase; primase; replication.
Figures
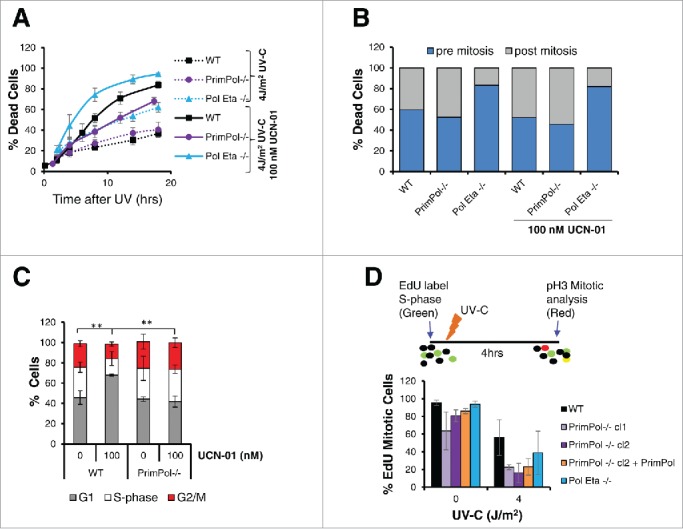
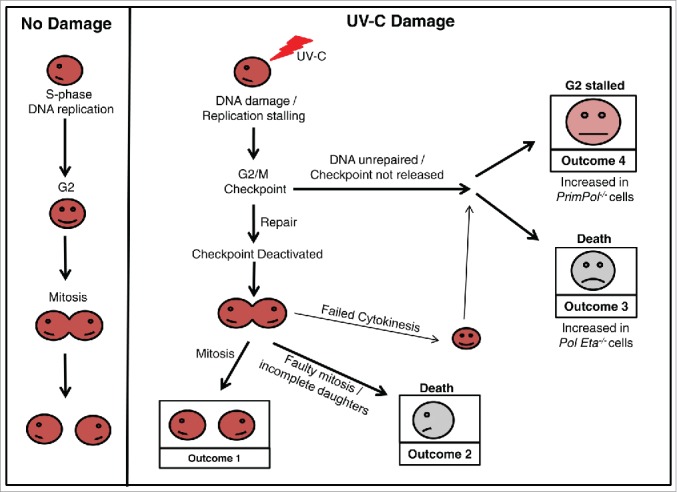
References
-
- Batista LF, Kaina B, Meneghini R, Menck CF. How DNA lesions are turned into powerful killing structures: insights from UV-induced apoptosis. Mutat Res 2009; 681:197-208; PMID:18845270; http://dx.doi.org/10.1016/j.mrrev.2008.09.001 - DOI - PubMed
-
- Arthanari H, Bolton PH. Functional and dysfunctional roles of quadruplex DNA in cells. Chem Biol 2001; 8:221-30; PMID:11306347; http://dx.doi.org/10.1016/S1074-5521(01)00007-2 - DOI - PubMed
-
- Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem 2005; 74:317-53; PMID:15952890; http://dx.doi.org/10.1146/annurev.biochem.74.082803.133250 - DOI - PubMed
-
- Sale JE, Lehmann AR, Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nature Reviews Mol Cell Biol 2012; 13:141-52; http://dx.doi.org/10.1038/nrm3289 - DOI - PMC - PubMed
-
- Livneh Z, Ziv O, Shachar S. Multiple two-polymerase mechanisms in mammalian translesion DNA synthesis. Cell Cycle 2010; 9:729-35; PMID:20139724; http://dx.doi.org/10.4161/cc.9.4.10727 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous