

The Role of Innate Immunity in Alcoholic Liver Disease
- PMID: 26695748
- PMCID: PMC4590620
- DOI: 10.35946/arcr.v37.2.08
The Role of Innate Immunity in Alcoholic Liver Disease
Abstract
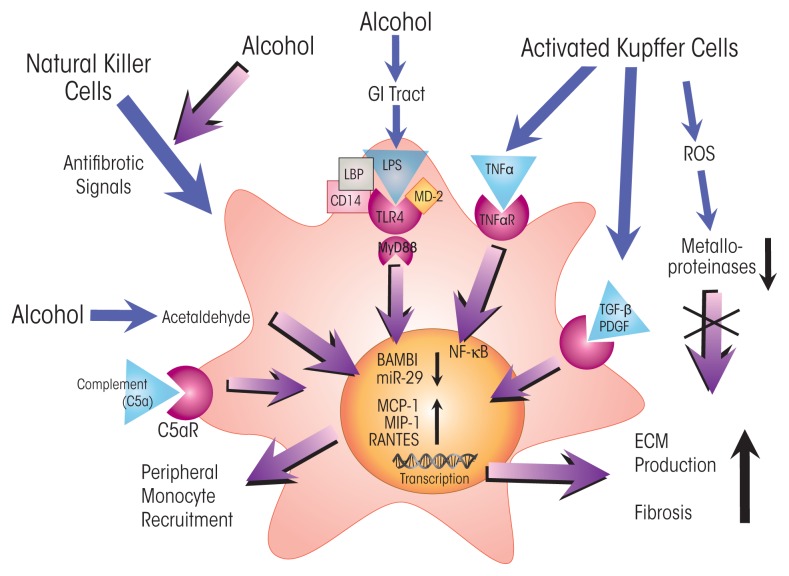
The innate immune system represents the first-line response to invading microbes, tissue damage, or aberrant cell growth. Many of the proteins and cells involved in innate immunity are produced by, and reside in, the liver. This abundance in immune cells and proteins reflects the liver's adaptation to various immune challenges but also makes the organ particularly vulnerable to alcohol's effects. Heavy alcohol consumption may produce leakage of microbes and microbial products from the gastrointestinal tract, which quickly reach the liver via the portal vein. Exposure to these immune challenges and to alcohol and its breakdown products dysregulates the liver's normally fine-tuned immune signaling pathways, leading to activation of various cellular sensors of pathogen- or damage-associated molecular patterns. The ensuing expression of pro-inflammatory cytokines (e.g., tumor necrosis factor a [TNFα], interleukin [IL]-8, and IL-1b) results in cellular dysfunction that contributes to alcoholic liver disease (ALD). Investigations into the roles of the various components of liver innate immunity in ALD have begun to uncover the molecular basis of this disease. Further progress in this area may help inform the development of interventions targeting the innate system to augment current treatments of ALD. These treatments could include antibodies against pro-inflammatory cytokines, use of anti-inflammatory cytokines, or suppression of alcohol-induced epigenetic regulators of innate immunity.
Figures



References
-
- An L, Wang X, Cederbaum AI. Cytokines in alcoholic liver disease. Archives of Toxicology. 2012;86(9):1337–1348. - PubMed
-
- Bode C, Kugler V, Bode JC. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. Journal of Hepatology. 1987;4(1):8–14. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
