

Origin-Dependent Inverted-Repeat Amplification: Tests of a Model for Inverted DNA Amplification
- PMID: 26700858
- PMCID: PMC4689423
- DOI: 10.1371/journal.pgen.1005699
Origin-Dependent Inverted-Repeat Amplification: Tests of a Model for Inverted DNA Amplification
Abstract
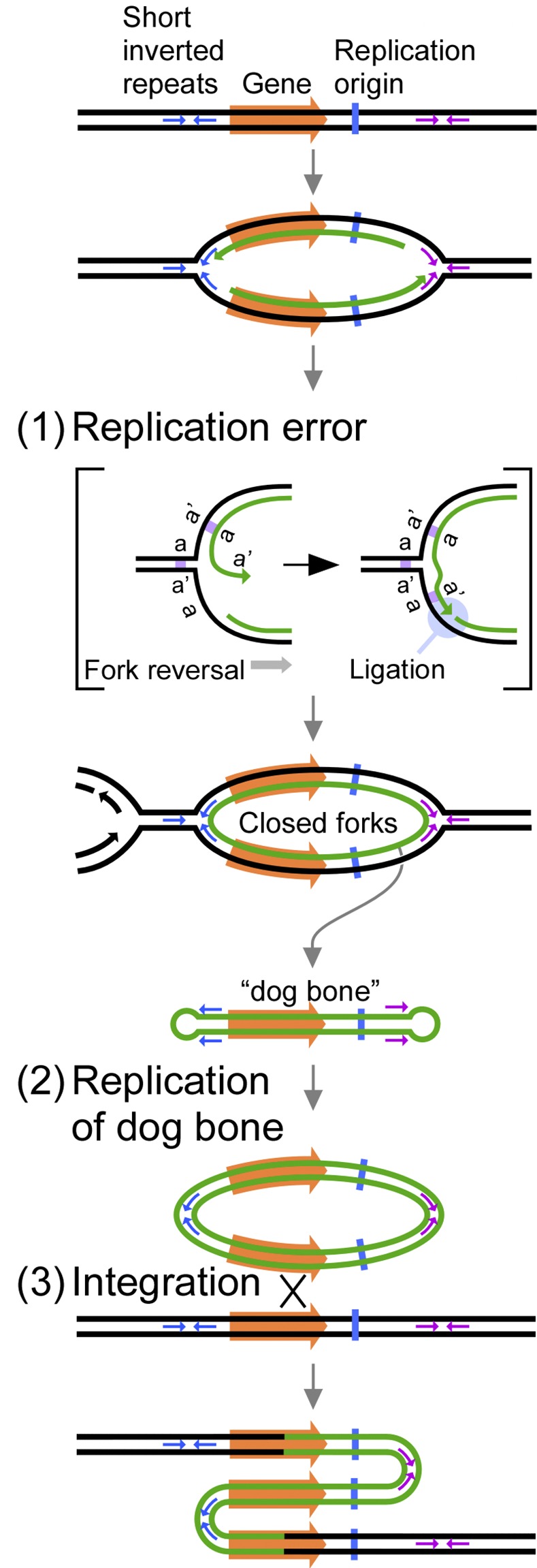
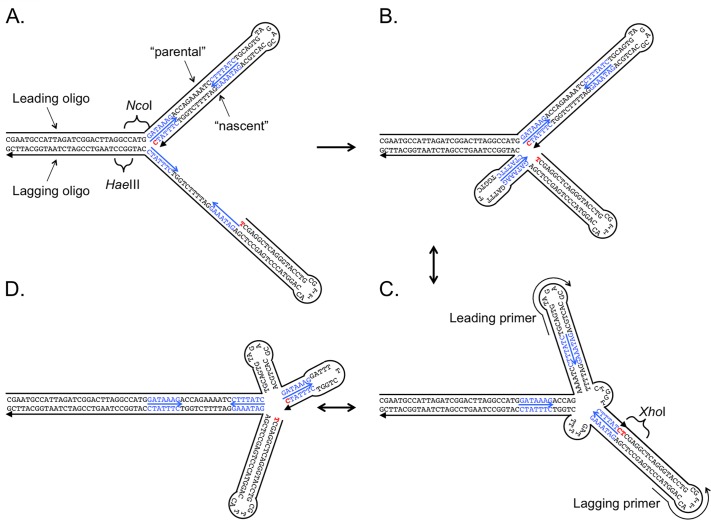
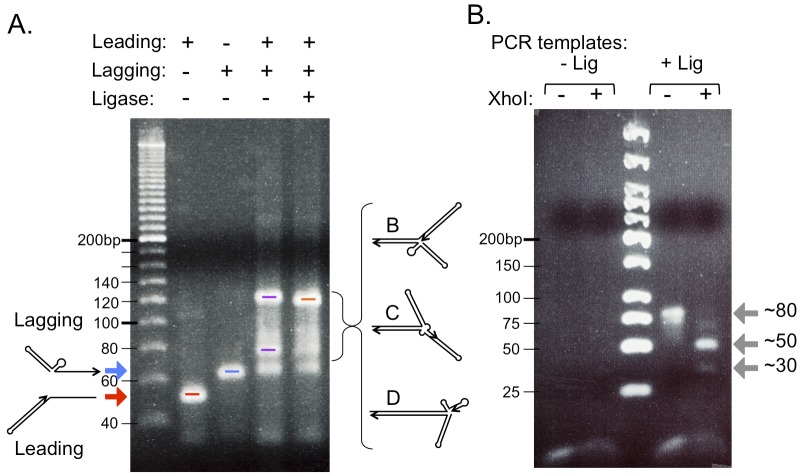
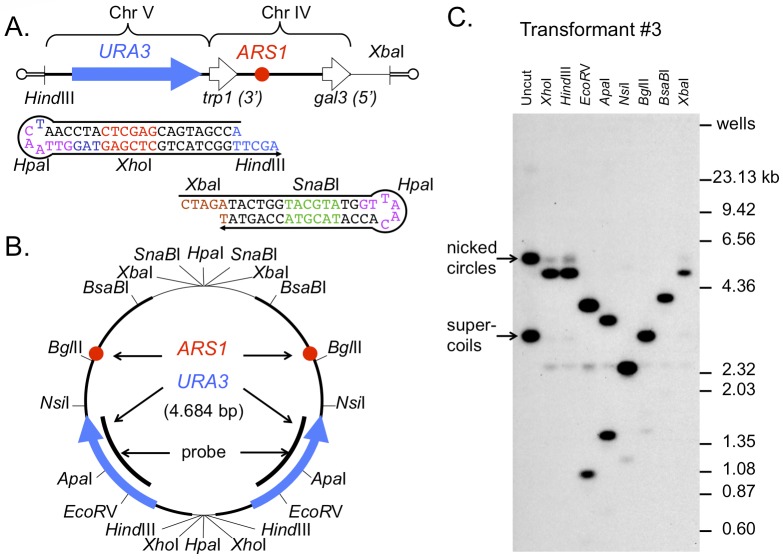
DNA replication errors are a major driver of evolution--from single nucleotide polymorphisms to large-scale copy number variations (CNVs). Here we test a specific replication-based model to explain the generation of interstitial, inverted triplications. While no genetic information is lost, the novel inversion junctions and increased copy number of the included sequences create the potential for adaptive phenotypes. The model--Origin-Dependent Inverted-Repeat Amplification (ODIRA)-proposes that a replication error at pre-existing short, interrupted, inverted repeats in genomic sequences generates an extrachromosomal, inverted dimeric, autonomously replicating intermediate; subsequent genomic integration of the dimer yields this class of CNV without loss of distal chromosomal sequences. We used a combination of in vitro and in vivo approaches to test the feasibility of the proposed replication error and its downstream consequences on chromosome structure in the yeast Saccharomyces cerevisiae. We show that the proposed replication error-the ligation of leading and lagging nascent strands to create "closed" forks-can occur in vitro at short, interrupted inverted repeats. The removal of molecules with two closed forks results in a hairpin-capped linear duplex that we show replicates in vivo to create an inverted, dimeric plasmid that subsequently integrates into the genome by homologous recombination, creating an inverted triplication. While other models have been proposed to explain inverted triplications and their derivatives, our model can also explain the generation of human, de novo, inverted amplicons that have a 2:1 mixture of sequences from both homologues of a single parent--a feature readily explained by a plasmid intermediate that arises from one homologue and integrates into the other homologue prior to meiosis. Our tests of key features of ODIRA lend support to this mechanism and suggest further avenues of enquiry to unravel the origins of interstitial, inverted CNVs pivotal in human health and evolution.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
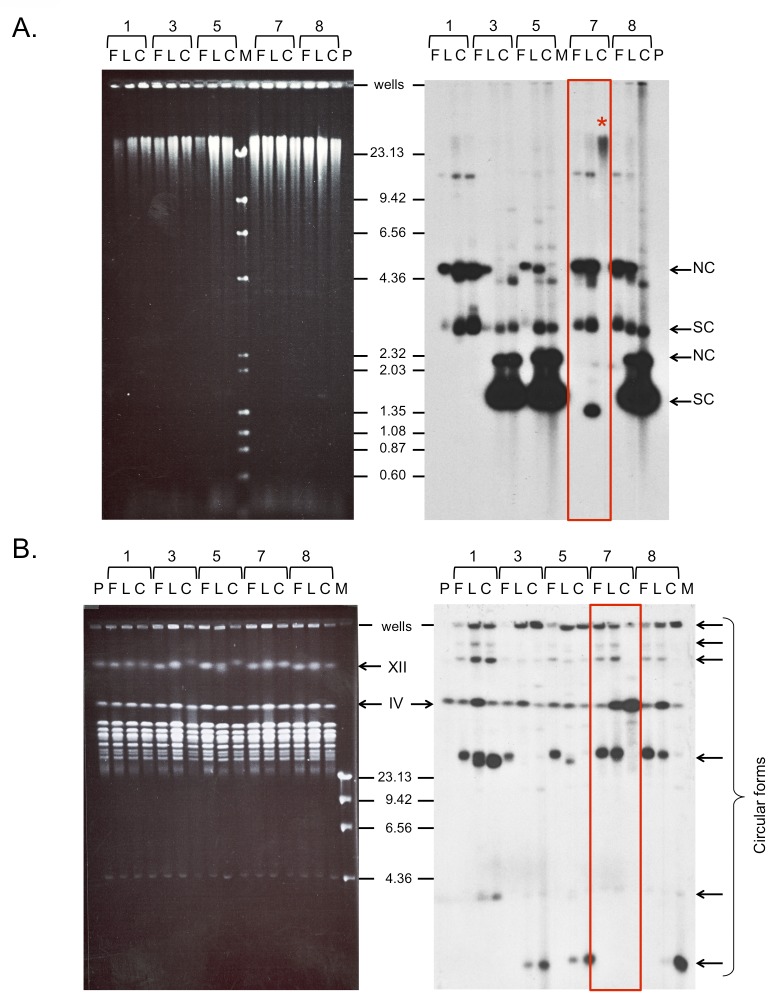
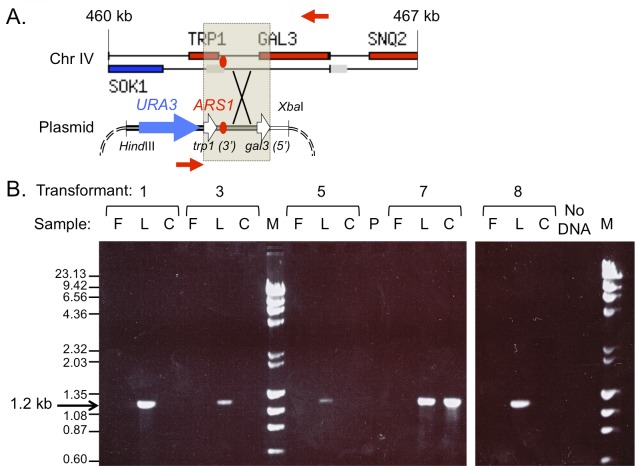
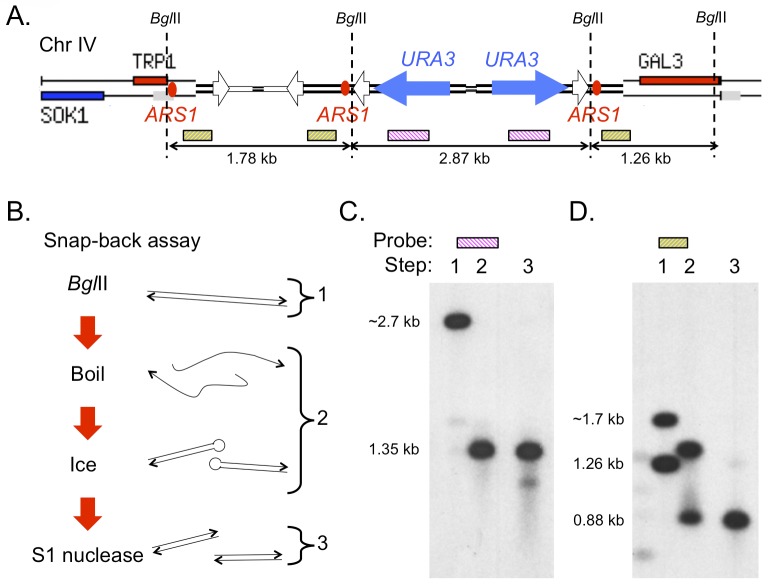
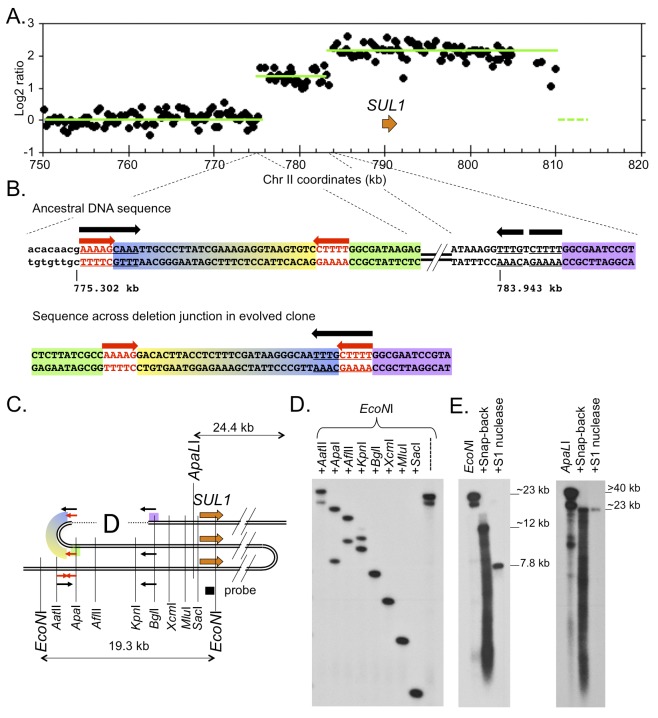
References
-
- Knijnenburg J, van Bever Y, Hulsman LO, van Kempen CA, Bolman GM, van Loon RL, et al. A 600 kb triplication in the cat eye syndrome critical region causes anorectal, renal and preauricular anomalies in a three-generation family. Eur J Hum Genet. 2012;20(9):986–9. 10.1038/ejhg.2012.43 - DOI - PMC - PubMed
Publication types
MeSH terms
Associated data
- BioProject/PRJNA291644
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
