

Autophagy: A Novel Therapeutic Target for Diabetic Nephropathy
- PMID: 26706914
- PMCID: PMC4696980
- DOI: 10.4093/dmj.2015.39.6.451
Autophagy: A Novel Therapeutic Target for Diabetic Nephropathy
Abstract
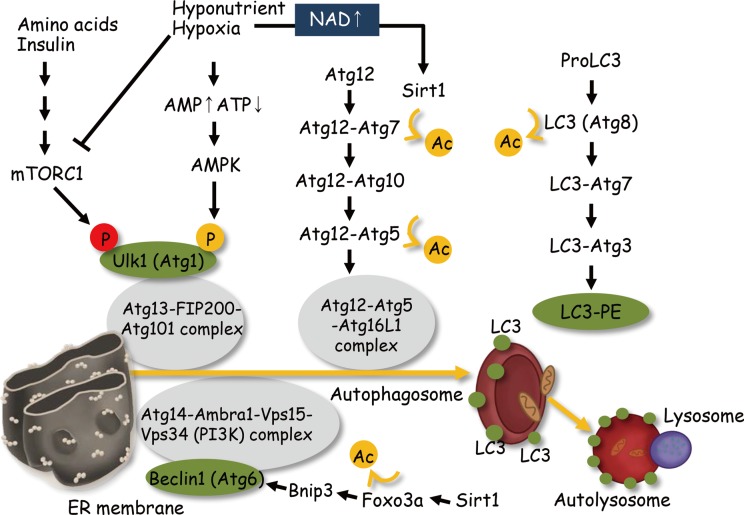
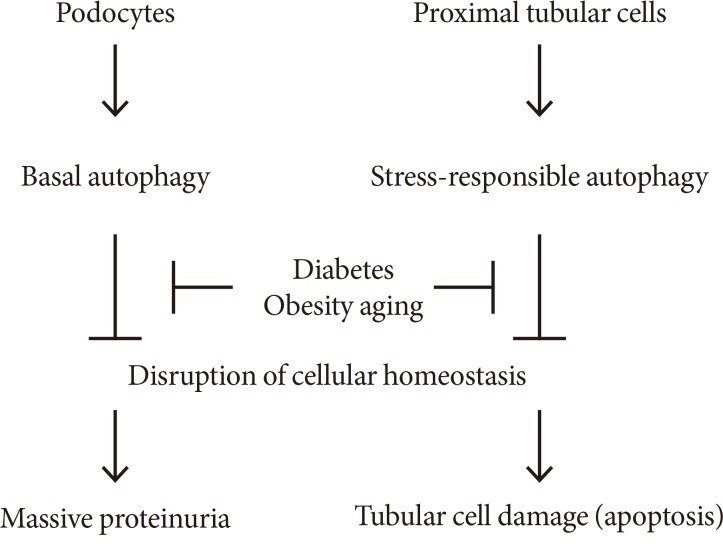
Diabetic nephropathy is a leading cause of end stage renal disease and its occurance is increasing worldwide. The most effective treatment strategy for the condition is intensive treatment to strictly control glycemia and blood pressure using renin-angiotensin system inhibitors. However, a fraction of patients still go on to reach end stage renal disease even under such intensive care. New therapeutic targets for diabetic nephropathy are, therefore, urgently needed. Autophagy is a major catabolic pathway by which mammalian cells degrade macromolecules and organelles to maintain intracellular homeostasis. The accumulation of damaged proteins and organelles is associated with the pathogenesis of diabetic nephropathy. Autophagy in the kidney is activated under some stress conditions, such as oxidative stress and hypoxia in proximal tubular cells, and occurs even under normal conditions in podocytes. These and other accumulating findings have led to a hypothesis that autophagy is involved in the pathogenesis of diabetic nephropathy. Here, we review recent findings underpinning this hypothesis and discuss the advantages of targeting autophagy for the treatment of diabetic nephropathy.
Keywords: AMP-activated protein kinases; Autophagy; Caloric restriction; Diabetic nephropathy; Mechanistic target of rapamycin complex 1; Podocytes; Sirt1; Tubular cell.
Conflict of interest statement
Figures

Similar articles
-
Autophagy: emerging therapeutic target for diabetic nephropathy.Semin Nephrol. 2014 Jan;34(1):9-16. doi: 10.1016/j.semnephrol.2013.11.003. Epub 2013 Nov 21. Semin Nephrol. 2014. PMID: 24485025 Review.
-
The role of autophagy in the pathogenesis of diabetic nephropathy.J Diabetes Res. 2013;2013:193757. doi: 10.1155/2013/193757. Epub 2013 Dec 17. J Diabetes Res. 2013. PMID: 24455746 Free PMC article. Review.
-
Regulating Autophagy as a Therapeutic Target for Diabetic Nephropathy.Curr Diab Rep. 2017 Jul;17(7):53. doi: 10.1007/s11892-017-0879-y. Curr Diab Rep. 2017. PMID: 28593583 Review.
-
Role of nutrient-sensing signals in the pathogenesis of diabetic nephropathy.Biomed Res Int. 2014;2014:315494. doi: 10.1155/2014/315494. Epub 2014 Jul 14. Biomed Res Int. 2014. PMID: 25126552 Free PMC article. Review.
-
[The latest advance of the relationship between autophagy and diabetic retinopathy].Zhonghua Yan Ke Za Zhi. 2012 Jul;48(7):649-52. Zhonghua Yan Ke Za Zhi. 2012. PMID: 22943871 Review. Chinese.
Cited by
-
Placenta-derived mesenchymal stem cells protect against diabetic kidney disease by upregulating autophagy-mediated SIRT1/FOXO1 pathway.Ren Fail. 2024 Dec;46(1):2303396. doi: 10.1080/0886022X.2024.2303396. Epub 2024 Jan 17. Ren Fail. 2024. PMID: 38234193 Free PMC article.
-
Nephroprotective potential of syringic acid in experimental diabetic nephropathy: Focus on oxidative stress and autophagy.Indian J Pharmacol. 2023 Jan-Feb;55(1):34-42. doi: 10.4103/ijp.ijp_671_22. Indian J Pharmacol. 2023. PMID: 36960519 Free PMC article.
-
P2Y2R contributes to the development of diabetic nephropathy by inhibiting autophagy response.Mol Metab. 2020 Dec;42:101089. doi: 10.1016/j.molmet.2020.101089. Epub 2020 Sep 25. Mol Metab. 2020. PMID: 32987187 Free PMC article.
-
Dictyophora Polysaccharide Attenuates As-Mediated PINK1/Parkin Pathway-Induced Mitophagy in L-02 Cell through Scavenging ROS.Molecules. 2022 Apr 28;27(9):2806. doi: 10.3390/molecules27092806. Molecules. 2022. PMID: 35566158 Free PMC article.
-
Role of sirtuin-1 in diabetic nephropathy.J Mol Med (Berl). 2019 Mar;97(3):291-309. doi: 10.1007/s00109-019-01743-7. Epub 2019 Feb 1. J Mol Med (Berl). 2019. PMID: 30707256 Free PMC article. Review.
References
-
- Abbate M, Zoja C, Remuzzi G. How does proteinuria cause progressive renal damage? J Am Soc Nephrol. 2006;17:2974–2984. - PubMed
-
- Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. - PubMed
-
- Dunlop M. Aldose reductase and the role of the polyol pathway in diabetic nephropathy. Kidney Int Suppl. 2000;77:S3–S12. - PubMed
-
- Forbes JM, Thallas V, Thomas MC, Founds HW, Burns WC, Jerums G, Cooper ME. The breakdown of preexisting advanced glycation end products is associated with reduced renal fibrosis in experimental diabetes. FASEB J. 2003;17:1762–1764. - PubMed
-
- Ha H, Hwang IA, Park JH, Lee HB. Role of reactive oxygen species in the pathogenesis of diabetic nephropathy. Diabetes Res Clin Pract. 2008;82(Suppl 1):S42–S45. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
