

Spinal muscular atrophy with respiratory distress type 1 (SMARD1) Report of a Spanish case with extended clinicopathological follow-up
- PMID: 26709713
- PMCID: PMC4806405
- DOI: 10.5414/NP300902
Spinal muscular atrophy with respiratory distress type 1 (SMARD1) Report of a Spanish case with extended clinicopathological follow-up
Abstract
Background: Spinal muscular atrophy with respiratory distress type 1 (SMARD1) is a clinically and genetically distinct and uncommon variant of SMA that results from irreversible degeneration of α-motor neurons in the anterior horns of the spinal cord and in ganglion cells on the spinal root ganglia.
Aims: To describe the clinical, electrophysiological, neuropathological, and genetic findings, at different stages from birth to death, of a Spanish child diagnosed with SMARD1.
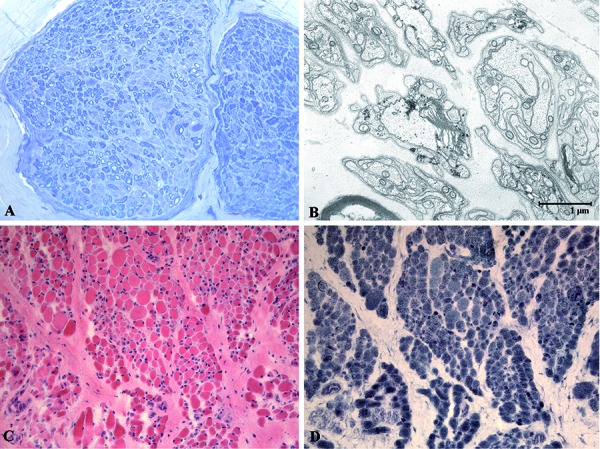
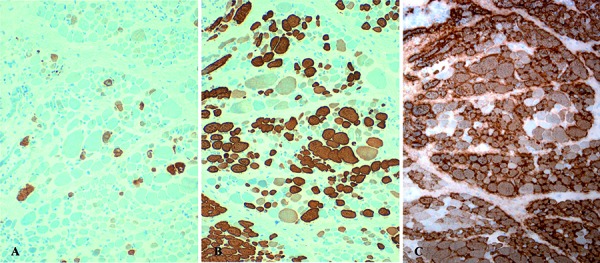
Patient and methods: We report the case of a 3-monthold girl with severe respiratory insufficiency and, later, intense hypotonia. Paraclinical tests included biochemistry, chest X-ray, and electrophysiological studies, among others. Muscle and nerve biopsies were performed at 5 and 10 months and studied under light and electron microscopy. Post-mortem examination and genetic investigations were performed.
Results: Pre- and post-mortem histopathological findings demonstrated the disease progression over time. Muscle biopsy at 5 months of age was normal, however a marked neurogenic atrophy was present in post-mortem samples. Peripheral motor and sensory nerves were severely involved likely due to a primary axonal disorder. Automatic sequencing of IGHMBP2 revealed a compound heterozygous mutation.
Conclusions: The diagnosis of SMARD1 should be considered in children with early respiratory insufficiency or in cases of atypical SMA. Direct sequencing of the IGHMBP2 gene should be performed.
Figures

References
-
- Mellins RB Hays AP Gold AP Berdon WE Bowdler JD Respiratory distress as the initial manifestation of Werdnig-Hoffmann disease. Pediatrics. 1974; 53: 33–40. - PubMed
-
- Grohmann K Wienker TF Saar K Rudnik-Schöneborn S Stoltenburg-Didinger G Rossi R Novelli G Nürnberg G Pfeufer A Wirth B Reis A Zerres K Hübner C Diaphragmatic spinal muscular atrophy with respiratory distress is heterogeneous, and one form Is linked to chromosome 11q13-q21. Am J Hum Genet. 1999; 65: 1459–1462. - PMC - PubMed
-
- Grohmann K Schuelke M Diers A Hoffmann K Lucke B Adams C Bertini E Leonhardt-Horti H Muntoni F Ouvrier R Pfeufer A Rossi R Van Maldergem L Wilmshurst JM Wienker TF Sendtner M Rudnik-Schöneborn S Zerres K Hübner C Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat Genet. 2001; 29: 75–77. - PubMed
-
- Grohmann K Varon R Stolz P Schuelke M Janetzki C Bertini E Bushby K Muntoni F Ouvrier R Van Maldergem L Goemans NM Lochmüller H Eichholz S Adams C Bosch F Grattan-Smith P Navarro C Neitzel H Polster T Topaloğlu H Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Ann Neurol. 2003; 54: 719–724. - PubMed
-
- Guenther UP Schuelke M Bertini E D’Amico A Goemans N Grohmann K Hübner C Varon R Genomic rearrangements at the IGHMBP2 gene locus in two patients with SMARD1. Hum Genet. 2004; 115: 319–326. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical