

A Next Generation Multiscale View of Inborn Errors of Metabolism
- PMID: 26712461
- PMCID: PMC4715559
- DOI: 10.1016/j.cmet.2015.11.012
A Next Generation Multiscale View of Inborn Errors of Metabolism
Abstract
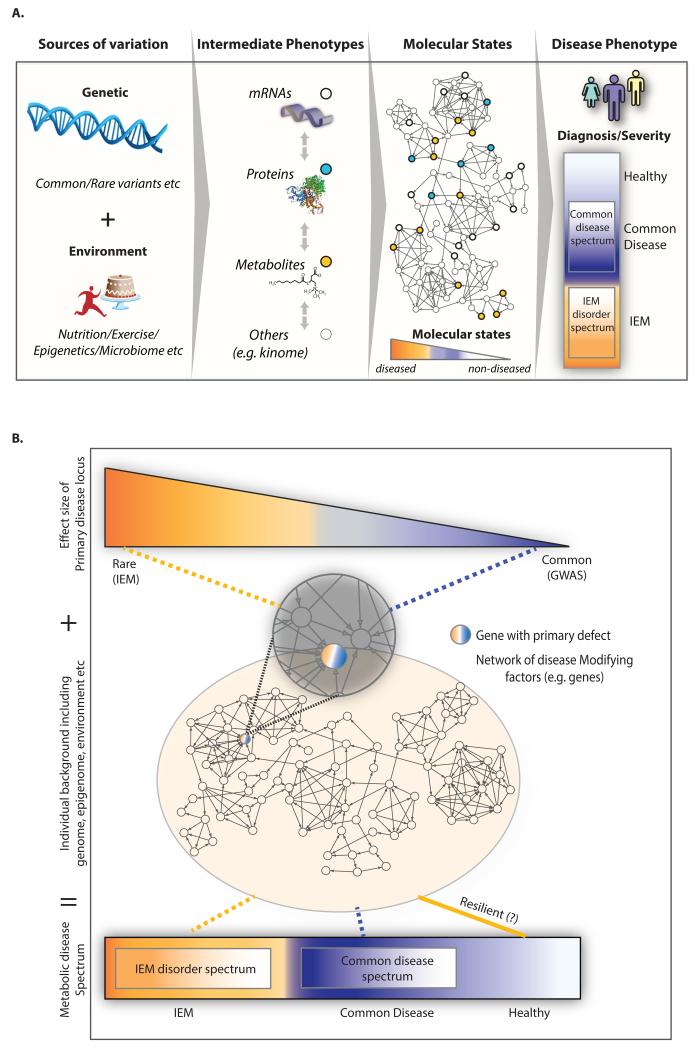
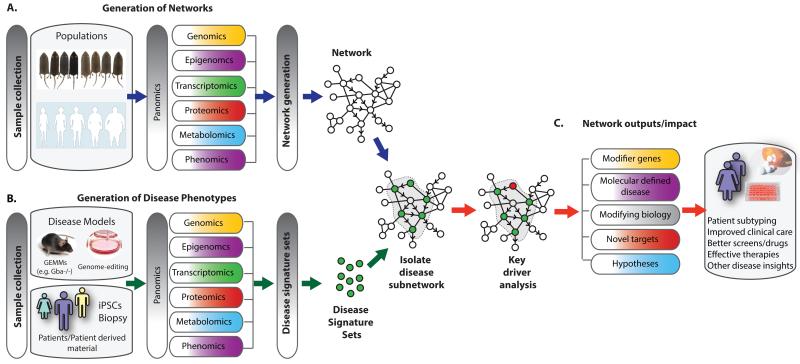
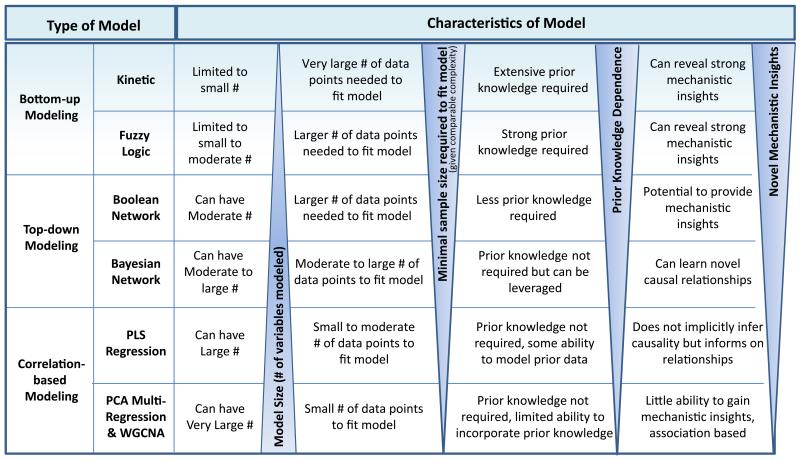
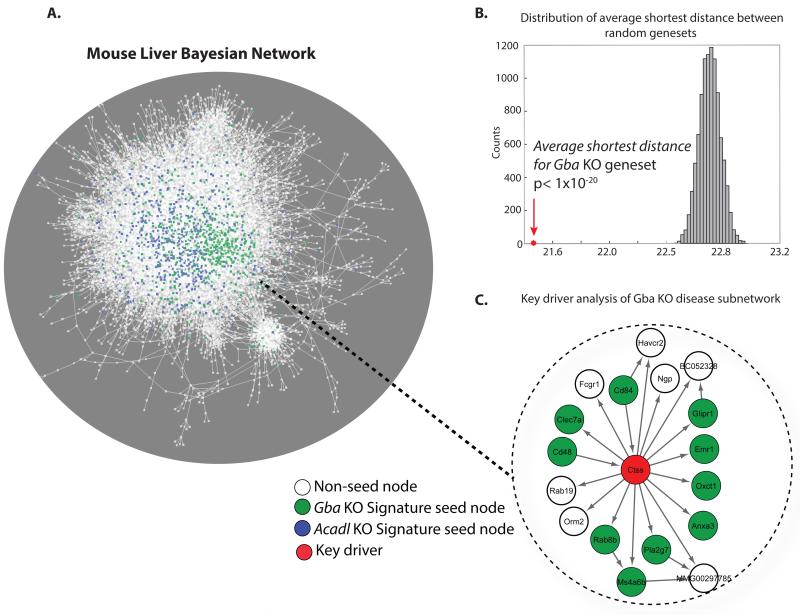
Inborn errors of metabolism (IEM) are not unlike common diseases. They often present as a spectrum of disease phenotypes that correlates poorly with the severity of the disease-causing mutations. This greatly impacts patient care and reveals fundamental gaps in our knowledge of disease modifying biology. Systems biology approaches that integrate multi-omics data into molecular networks have significantly improved our understanding of complex diseases. Similar approaches to study IEM are rare despite their complex nature. We highlight that existing common disease-derived datasets and networks can be repurposed to generate novel mechanistic insight in IEM and potentially identify candidate modifiers. While understanding disease pathophysiology will advance the IEM field, the ultimate goal should be to understand per individual how their phenotype emerges given their primary mutation on the background of their whole genome, not unlike personalized medicine. We foresee that panomics and network strategies combined with recent experimental innovations will facilitate this.
Keywords: human genetic disease; metabolism; network biology; omics.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
References
-
- Ala A, Schilsky M. Genetic modifiers of liver injury in hereditary liver disease. Semin Liver Dis. 2011;31:208–214. - PubMed
-
- Albert R, Thakar J. Boolean modeling: a logic-based dynamic approach for understanding signaling and regulatory networks and for making useful predictions. Wiley interdisciplinary reviews Systems biology and medicine. 2014;6:353–369. - PubMed
-
- Alfonso P, Navascues J, Navarro S, Medina P, Bolado-Carrancio A, Andreu V, Irun P, Rodriguez-Rey JC, Pocovi M, Espana F, et al. Characterization of variants in the glucosylceramide synthase gene and their association with type 1 Gaucher disease severity. Human mutation. 2013;34:1396–1403. - PubMed
-
- Andresen BS, Dobrowolski SF, O’Reilly L, Muenzer J, McCandless SE, Frazier DM, Udvari S, Bross P, Knudsen I, Banas R, et al. Medium-chain acyl-CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am J Hum Genet. 2001;68:1408–1418. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
