

Hypoxia Increases IGFBP-1 Phosphorylation Mediated by mTOR Inhibition
- PMID: 26714229
- PMCID: PMC4792228
- DOI: 10.1210/me.2015-1194
Hypoxia Increases IGFBP-1 Phosphorylation Mediated by mTOR Inhibition
Abstract
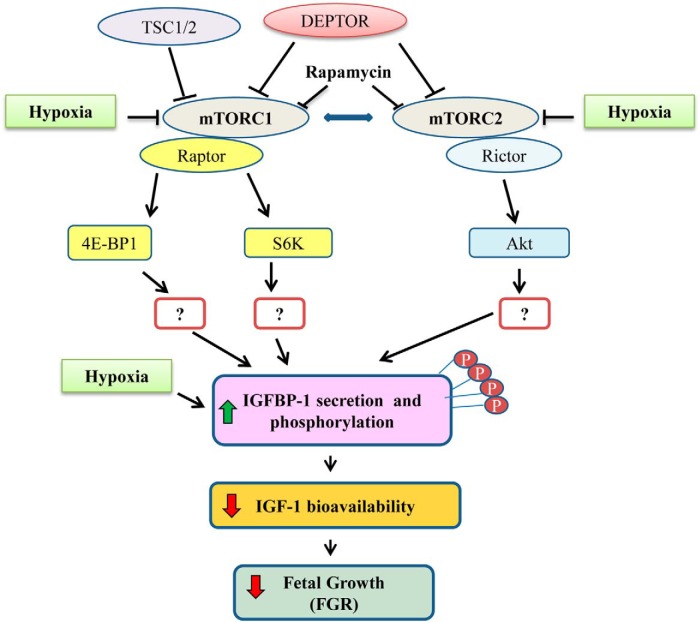
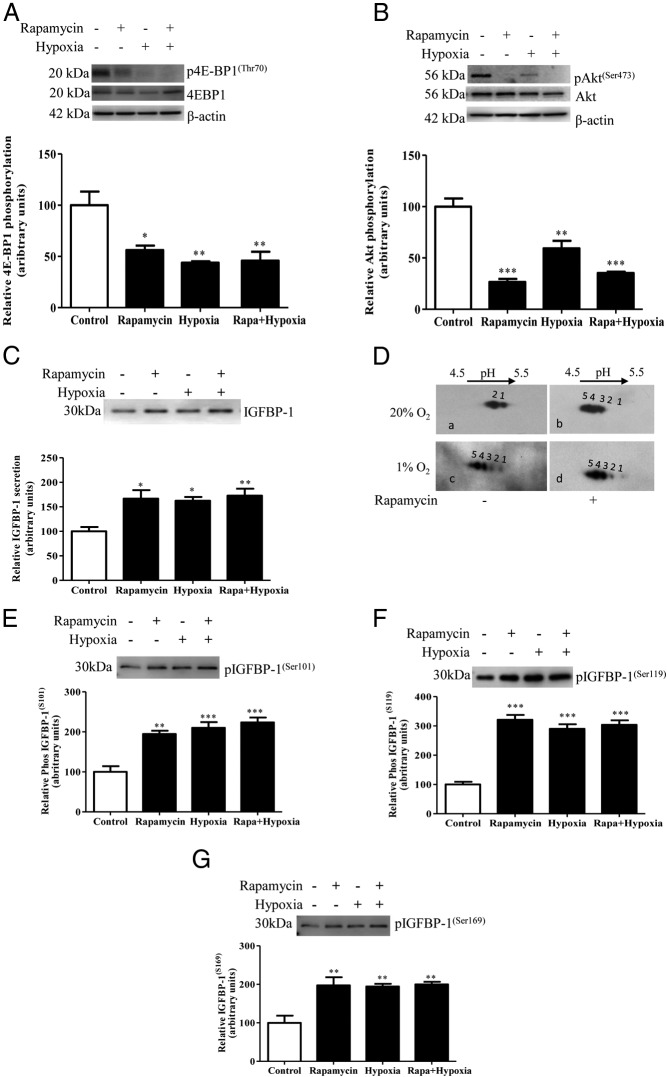
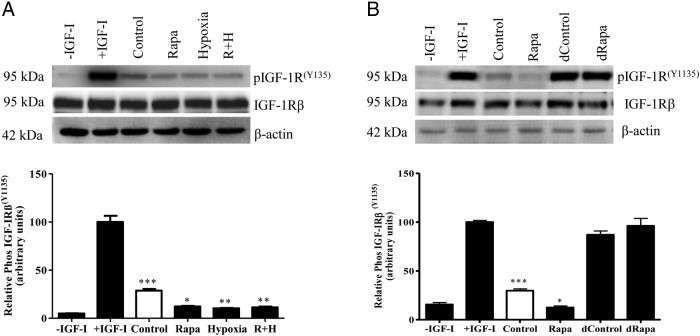
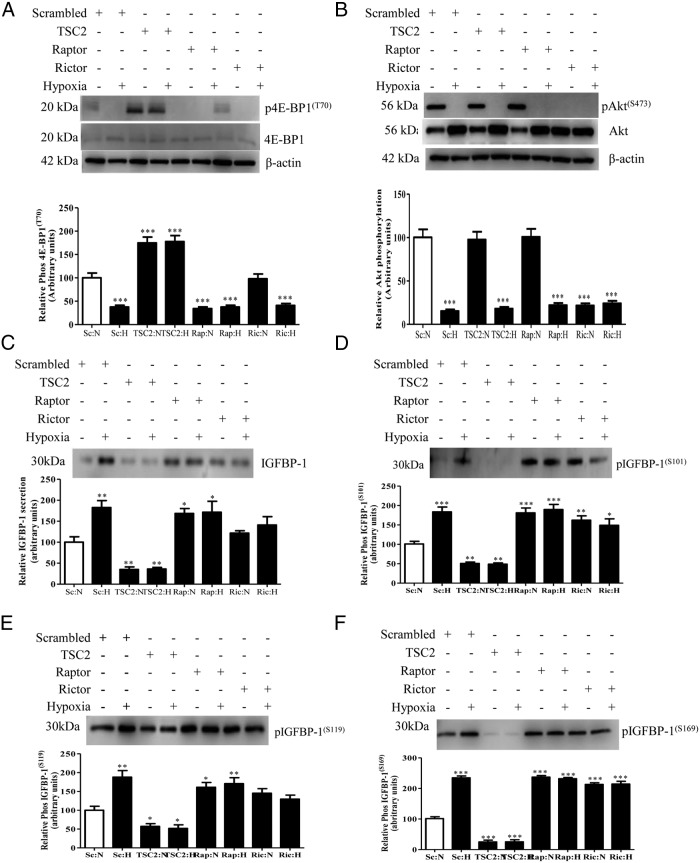
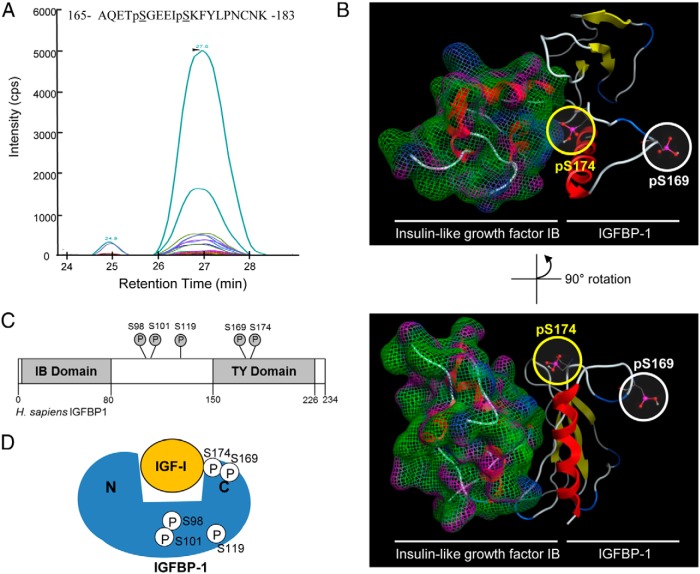
In fetal growth restriction (FGR), fetal growth is limited by reduced nutrient and oxygen supply. Insulin-like growth factor I (IGF-I) is a key regulator of fetal growth and IGF binding protein -1(IGFBP-1) is the principal regulator of fetal IGF-I bioavailability. Phosphorylation enhances IGFBP-1's affinity for IGF-I. Hypoxia induces IGFBP-1 hyperphosphorylation, markedly decreasing IGF-I bioavailability. We recently reported that fetal liver IGFBP-1 hyperphosphorylation is associated with inhibition of the mechanistic target of rapamycin (mTOR) in a nonhuman primate model of FGR. Here, we test the hypothesis that IGFBP-1 hyperphosphorylation in response to hypoxia is mediated by mTOR inhibition. We inhibited mTOR either by rapamycin or small interfering RNA (siRNA) targeting raptor (mTOR complex [mTORC]1) and/or rictor (mTORC2) in HepG2 cells cultured under hypoxia (1% O2) or basal (20% O2) conditions. Conversely, we activated mTORC1 or mTORC1+mTORC2 by silencing endogenous mTOR inhibitors (tuberous sclerosis complex 2/DEP-domain-containing and mTOR-interacting protein). Immunoblot analysis demonstrated that both hypoxia and inhibition of mTORC1 and/or mTORC2 induced similar degrees of IGFBP-1 phosphorylation at Ser101/119/169 and reduced IGF-I receptor autophosphorylation. Activation of mTORC1+mTORC2 or mTORC1 alone prevented IGFBP-1 hyperphosphorylation in response to hypoxia. Multiple reaction monitoring-mass spectrometry showed that rapamycin and/or hypoxia increased phosphorylation also at Ser98 and at a novel site Ser174. In silico structural analysis indicated that Ser174 was in close proximity to the IGF-binding site. Together, we demonstrate that signaling through the mTORC1 or mTORC2 pathway is sufficient to induce IGFBP-1 hyperphosphorylation in response to hypoxia. This study provides novel understanding of the cellular mechanism that controls fetal IGFBP-1 phosphorylation in hypoxia, and we propose that mTOR inhibition constitutes a mechanistic link between hypoxia, reduced IGF-I bioavailability and FGR.
Figures


Similar articles
-
Placental Remote Control of Fetal Metabolism: Trophoblast mTOR Signaling Regulates Liver IGFBP-1 Phosphorylation and IGF-1 Bioavailability.Int J Mol Sci. 2023 Apr 14;24(8):7273. doi: 10.3390/ijms24087273. Int J Mol Sci. 2023. PMID: 37108437 Free PMC article.
-
Liver mTOR controls IGF-I bioavailability by regulation of protein kinase CK2 and IGFBP-1 phosphorylation in fetal growth restriction.Endocrinology. 2014 Apr;155(4):1327-39. doi: 10.1210/en.2013-1759. Epub 2014 Jan 17. Endocrinology. 2014. PMID: 24437487 Free PMC article.
-
IGFBP-1 hyperphosphorylation in response to leucine deprivation is mediated by the AAR pathway.Mol Cell Endocrinol. 2015 Sep 5;412:182-95. doi: 10.1016/j.mce.2015.04.031. Epub 2015 May 5. Mol Cell Endocrinol. 2015. PMID: 25957086 Free PMC article.
-
The Role of Mammalian Target of Rapamycin (mTOR) in Insulin Signaling.Nutrients. 2017 Oct 27;9(11):1176. doi: 10.3390/nu9111176. Nutrients. 2017. PMID: 29077002 Free PMC article. Review.
-
Targeted Inhibition of Rictor/mTORC2 in Cancer Treatment: A New Era after Rapamycin.Curr Cancer Drug Targets. 2016;16(4):288-304. doi: 10.2174/1568009616666151113120830. Curr Cancer Drug Targets. 2016. PMID: 26563881 Review.
Cited by
-
Therapeutic relevance of mTOR inhibition in murine succinate semialdehyde dehydrogenase deficiency (SSADHD), a disorder of GABA metabolism.Biochim Biophys Acta Mol Basis Dis. 2017 Jan;1863(1):33-42. doi: 10.1016/j.bbadis.2016.10.009. Epub 2016 Oct 17. Biochim Biophys Acta Mol Basis Dis. 2017. PMID: 27760377 Free PMC article.
-
IUGR Is Associated With Marked Hyperphosphorylation of Decidual and Maternal Plasma IGFBP-1.J Clin Endocrinol Metab. 2019 Feb 1;104(2):408-422. doi: 10.1210/jc.2018-00820. J Clin Endocrinol Metab. 2019. PMID: 30124960 Free PMC article.
-
Novel roles of mechanistic target of rapamycin signaling in regulating fetal growth†.Biol Reprod. 2019 Apr 1;100(4):872-884. doi: 10.1093/biolre/ioy249. Biol Reprod. 2019. PMID: 30476008 Free PMC article. Review.
-
Pre-Conditioning with IFN-γ and Hypoxia Enhances the Angiogenic Potential of iPSC-Derived MSC Secretome.Cells. 2022 Mar 14;11(6):988. doi: 10.3390/cells11060988. Cells. 2022. PMID: 35326438 Free PMC article.
-
Placental Remote Control of Fetal Metabolism: Trophoblast mTOR Signaling Regulates Liver IGFBP-1 Phosphorylation and IGF-1 Bioavailability.Int J Mol Sci. 2023 Apr 14;24(8):7273. doi: 10.3390/ijms24087273. Int J Mol Sci. 2023. PMID: 37108437 Free PMC article.
References
-
- Eleftheriades M, Creatsas G, Nicolaides K. Fetal growth restriction and postnatal development. Ann NY Acad Sci. 2006;1092:319–330. - PubMed
-
- Kinzler WL, Vintzileos AM. Fetal growth restriction: a modern approach. Curr Opin Obstet Gynecol. 2008;20:125–131. - PubMed
-
- Ruijtenbeek K, Kessels LC, De Mey JG, Blanco CE. Chronic moderate hypoxia and protein malnutrition both induce growth retardation, but have distinct effects on arterial endothelium-dependent reactivity in the chicken embryo. Pediatr Res. 2003;53:573–579. - PubMed
-
- Randhawa R, Cohen P. The role of the insulin-like growth factor system in prenatal growth. Mol Genet Metab. 2005;86:84–90. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
