

Pathosphere.org: pathogen detection and characterization through a web-based, open source informatics platform
- PMID: 26714571
- PMCID: PMC4696252
- DOI: 10.1186/s12859-015-0840-5
Pathosphere.org: pathogen detection and characterization through a web-based, open source informatics platform
Abstract
Background: The detection of pathogens in complex sample backgrounds has been revolutionized by wide access to next-generation sequencing (NGS) platforms. However, analytical methods to support NGS platforms are not as uniformly available. Pathosphere (found at Pathosphere.org) is a cloud - based open - sourced community tool that allows for communication, collaboration and sharing of NGS analytical tools and data amongst scientists working in academia, industry and government. The architecture allows for users to upload data and run available bioinformatics pipelines without the need for onsite processing hardware or technical support.
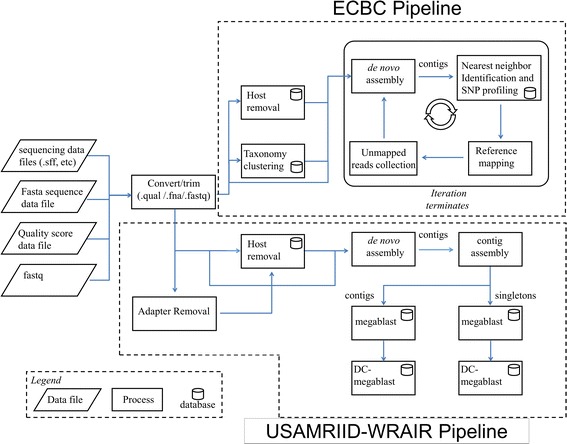
Results: The pathogen detection capabilities hosted on Pathosphere were tested by analyzing pathogen-containing samples sequenced by NGS with both spiked human samples as well as human and zoonotic host backgrounds. Pathosphere analytical pipelines developed by Edgewood Chemical Biological Center (ECBC) identified spiked pathogens within a common sample analyzed by 454, Ion Torrent, and Illumina sequencing platforms. ECBC pipelines also correctly identified pathogens in human samples containing arenavirus in addition to animal samples containing flavivirus and coronavirus. These analytical methods were limited in the detection of sequences with limited homology to previous annotations within NCBI databases, such as parvovirus. Utilizing the pipeline-hosting adaptability of Pathosphere, the analytical suite was supplemented by analytical pipelines designed by the United States Army Medical Research Insititute of Infectious Diseases and Walter Reed Army Institute of Research (USAMRIID-WRAIR). These pipelines were implemented and detected parvovirus sequence in the sample that the ECBC iterative analysis previously failed to identify.
Conclusions: By accurately detecting pathogens in a variety of samples, this work demonstrates the utility of Pathosphere and provides a platform for utilizing, modifying and creating pipelines for a variety of NGS technologies developed to detect pathogens in complex sample backgrounds. These results serve as an exhibition for the existing pipelines and web-based interface of Pathosphere as well as the plug-in adaptability that allows for integration of newer NGS analytical software as it becomes available.
Figures

Similar articles
-
DolphinNext: a distributed data processing platform for high throughput genomics.BMC Genomics. 2020 Apr 19;21(1):310. doi: 10.1186/s12864-020-6714-x. BMC Genomics. 2020. PMID: 32306927 Free PMC article.
-
DNAscan: personal computer compatible NGS analysis, annotation and visualisation.BMC Bioinformatics. 2019 Apr 27;20(1):213. doi: 10.1186/s12859-019-2791-8. BMC Bioinformatics. 2019. PMID: 31029080 Free PMC article.
-
Bio-Docklets: virtualization containers for single-step execution of NGS pipelines.Gigascience. 2017 Aug 1;6(8):1-7. doi: 10.1093/gigascience/gix048. Gigascience. 2017. PMID: 28854616 Free PMC article.
-
OpenContami: a web-based application for detecting microbial contaminants in next-generation sequencing data.Bioinformatics. 2021 Sep 29;37(18):3021-3022. doi: 10.1093/bioinformatics/btab101. Bioinformatics. 2021. PMID: 33576798 Free PMC article. Review.
-
OTP: An automatized system for managing and processing NGS data.J Biotechnol. 2017 Nov 10;261:53-62. doi: 10.1016/j.jbiotec.2017.08.006. Epub 2017 Aug 10. J Biotechnol. 2017. PMID: 28803971 Review.
Cited by
-
Targeted Sequencing of Respiratory Viruses in Clinical Specimens for Pathogen Identification and Genome-Wide Analysis.Methods Mol Biol. 2018;1838:125-140. doi: 10.1007/978-1-4939-8682-8_10. Methods Mol Biol. 2018. PMID: 30128994 Free PMC article.
-
Metagenomics for Clinical Infectious Disease Diagnostics Steps Closer to Reality.J Clin Microbiol. 2018 Aug 27;56(9):e00850-18. doi: 10.1128/JCM.00850-18. Print 2018 Sep. J Clin Microbiol. 2018. PMID: 29976592 Free PMC article.
-
Metagenome analysis of viruses associated with Anopheles mosquitoes from Ramu Upazila, Cox's Bazar District, Bangladesh.PeerJ. 2025 Mar 31;13:e19180. doi: 10.7717/peerj.19180. eCollection 2025. PeerJ. 2025. PMID: 40183042 Free PMC article.
-
Coding-Complete Genome Sequence and Phylogenetic Relatedness of a SARS-CoV-2 Strain Detected in March 2020 in Cameroon.Microbiol Resour Announc. 2021 Mar 11;10(10):e00093-21. doi: 10.1128/MRA.00093-21. Microbiol Resour Announc. 2021. PMID: 33707325 Free PMC article.
-
Metagenomic Analysis Reveals Three Novel and Prevalent Mosquito Viruses from a Single Pool of Aedes vexans nipponii Collected in the Republic of Korea.Viruses. 2019 Mar 5;11(3):222. doi: 10.3390/v11030222. Viruses. 2019. PMID: 30841520 Free PMC article.
References
-
- Leopold SR, Goering RV, Witten A, Harmsen D, Mellmann A. Bacterial whole genome sequencing revisited: portable, scalable and standardized analysis for typing and detection of virulence and antibiotic resistance genes. J Clin Microbiol. 2014;52:2365–70. doi: 10.1128/JCM.00262-14. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
