

Modulation of post-stroke degenerative and regenerative processes and subacute protection by site-targeted inhibition of the alternative pathway of complement
- PMID: 26714866
- PMCID: PMC4696299
- DOI: 10.1186/s12974-015-0464-8
Modulation of post-stroke degenerative and regenerative processes and subacute protection by site-targeted inhibition of the alternative pathway of complement
Abstract
Background: Complement promotes neuroinflammation and injury in models of stroke. However, complement is also being increasingly implicated in repair and regeneration after central nervous system (CNS) injury, and some complement deficiencies have been shown to provide acute, but not subacute, protection after murine stroke. Here, we investigate the dual role of complement in injury and repair after cerebral ischemia and reperfusion.
Methods: We used complement-deficient mice and different complement inhibitors in a model of transient middle cerebral artery occlusion to investigate complement-dependent cellular and molecular changes that occur through the subacute phase after stroke.
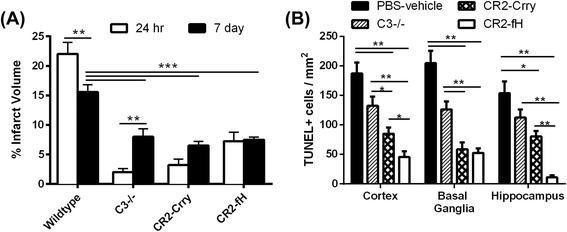
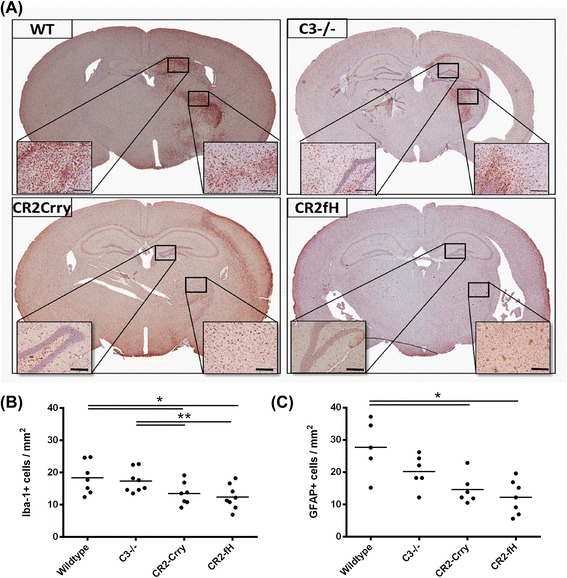
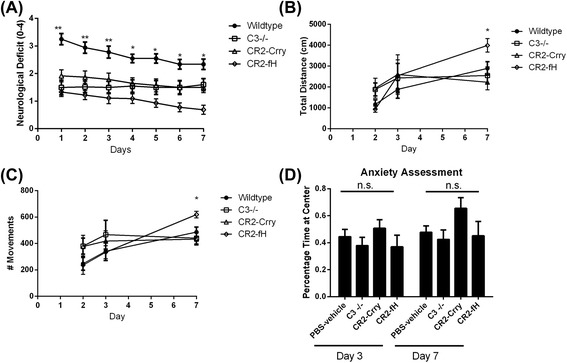
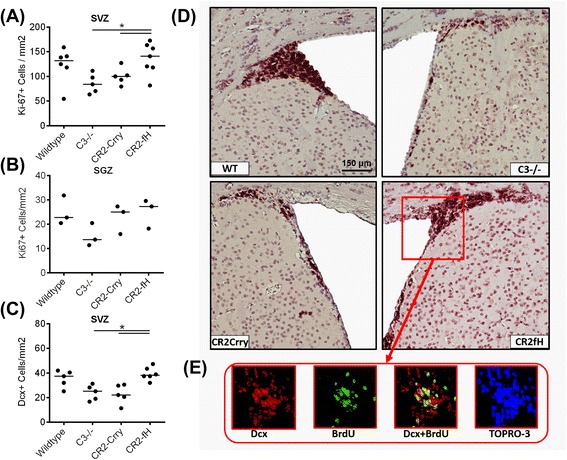
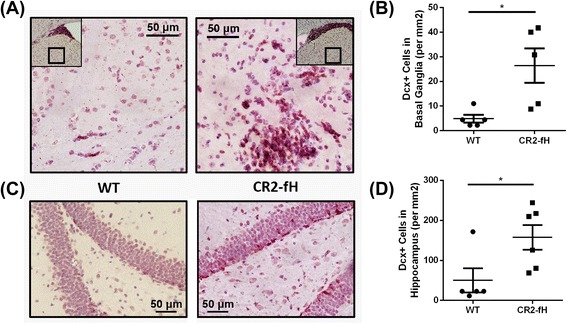
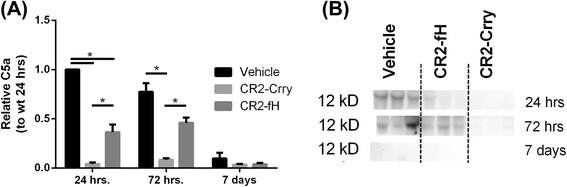
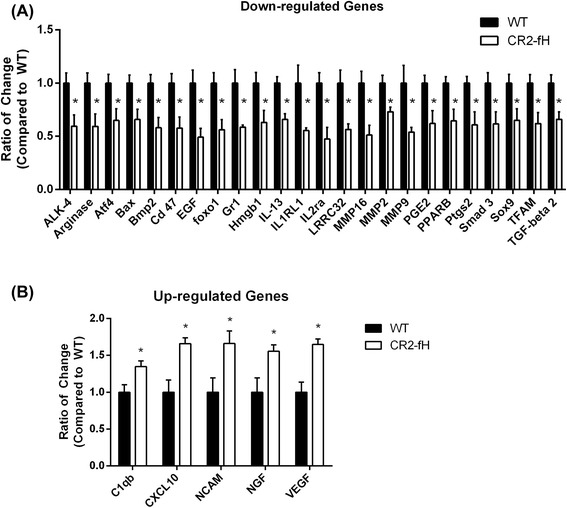
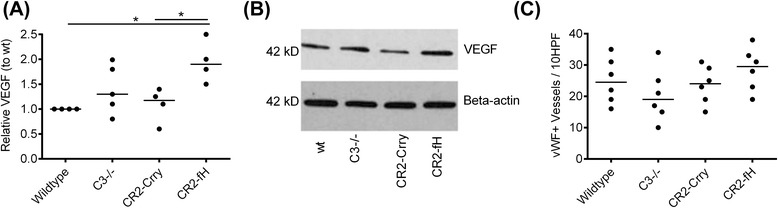
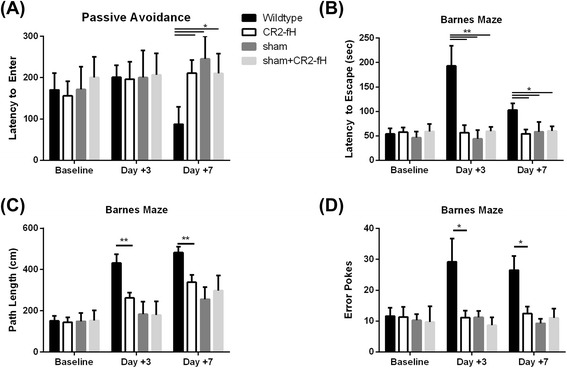
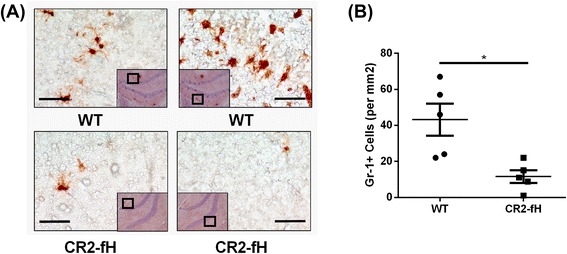
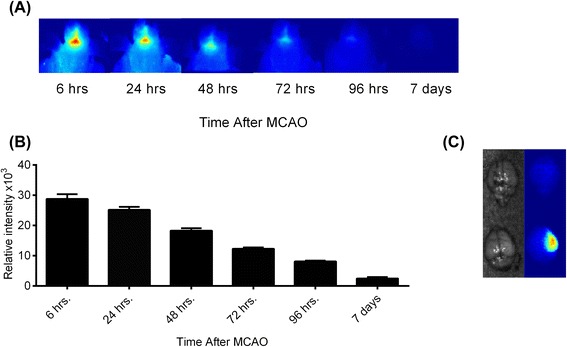
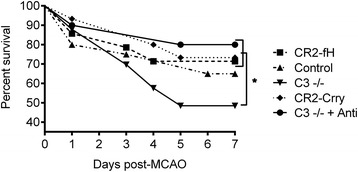
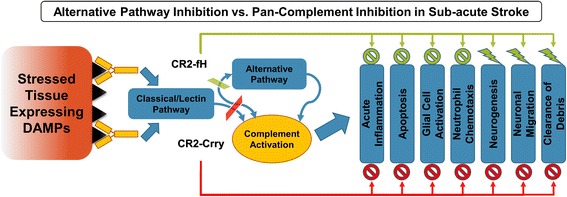
Results: C3 deficiency and site-targeted complement inhibition with either CR2-Crry (inhibits all pathways) or CR2-fH (inhibits alternative pathway) significantly reduced infarct size, reduced apoptotic cell death, and improved neurological deficit score in the acute phase after stroke. However, only in CR2-fH-treated mice was there sustained protection with no evolution of injury in the subacute phase. Whereas both inhibitors significantly reduced microglia/macrophage activation and astrogliosis in the subacute phase, only CR2-fH improved neurological deficit and locomotor function, maintained neurogenesis markers, enhanced neuronal migration, and increased VEGF expression. These findings in CR2-fH-treated mice correlated with improved performance in spatial learning and passive avoidance tasks. The complement anaphylatoxins have been implicated in repair and regenerative mechanisms after CNS injury, and in this context CR2-fH significantly reduced, but did not eliminate the generation of C5a within the brain, unlike CR2-Crry that completely blocked C5a generation. Gene expression profiling revealed that CR2-fH treatment downregulated genes associated with apoptosis, TGFβ signaling, and neutrophil activation, and decreased neutrophil infiltration was confirmed by immunohistochemistry. CR2-fH upregulated genes for neural growth factor and mediators of neurogenesis and neuronal migration. Live animal imaging demonstrated that following intravenous injection, CR2-fH targeted specifically to the post-ischemic brain, with a tissue half-life of 48.5 h. Finally, unlike C3 deficiency, targeted complement inhibition did not increase susceptibility to lethal post-stroke infection, an important consideration for stroke patients.
Conclusions: Ischemic brain tissue-targeted and selective inhibition of alternative complement pathway provide self-limiting inhibition of complement activation and reduces acute injury while maintaining complement-dependent recovery mechanisms into the subacute phase after stroke.
Figures
References
-
- Komotar RJ, Kim GH, Otten ML, Hassid B, Mocco J, Sughrue ME, et al. The role of complement in stroke therapy. Adv Exp Med Biol. 2008;632:23–33. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
