

A new strategy for enhancing imputation quality of rare variants from next-generation sequencing data via combining SNP and exome chip data
- PMID: 26715385
- PMCID: PMC4696174
- DOI: 10.1186/s12864-015-2192-y
A new strategy for enhancing imputation quality of rare variants from next-generation sequencing data via combining SNP and exome chip data
Abstract
Background: Rare variants have gathered increasing attention as a possible alternative source of missing heritability. Since next generation sequencing technology is not yet cost-effective for large-scale genomic studies, a widely used alternative approach is imputation. However, the imputation approach may be limited by the low accuracy of the imputed rare variants. To improve imputation accuracy of rare variants, various approaches have been suggested, including increasing the sample size of the reference panel, using sequencing data from study-specific samples (i.e., specific populations), and using local reference panels by genotyping or sequencing a subset of study samples. While these approaches mainly utilize reference panels, imputation accuracy of rare variants can also be increased by using exome chips containing rare variants. The exome chip contains 250 K rare variants selected from the discovered variants of about 12,000 sequenced samples. If exome chip data are available for previously genotyped samples, the combined approach using a genotype panel of merged data, including exome chips and SNP chips, should increase the imputation accuracy of rare variants.
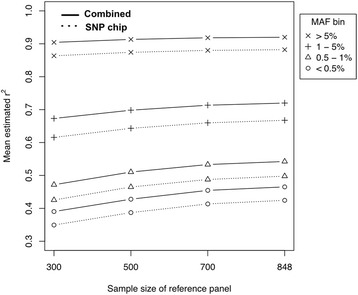
Results: In this study, we describe a combined imputation which uses both exome chip and SNP chip data simultaneously as a genotype panel. The effectiveness and performance of the combined approach was demonstrated using a reference panel of 848 samples constructed using exome sequencing data from the T2D-GENES consortium and 5,349 sample genotype panels consisting of an exome chip and SNP chip. As a result, the combined approach increased imputation quality up to 11 %, and genomic coverage for rare variants up to 117.7 % (MAF < 1 %), compared to imputation using the SNP chip alone. Also, we investigated the systematic effect of reference panels on imputation quality using five reference panels and three genotype panels. The best performing approach was the combination of the study specific reference panel and the genotype panel of combined data.
Conclusions: Our study demonstrates that combined datasets, including SNP chips and exome chips, enhances both the imputation quality and genomic coverage of rare variants.
Figures



References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
