

RAD Capture (Rapture): Flexible and Efficient Sequence-Based Genotyping
- PMID: 26715661
- PMCID: PMC4788223
- DOI: 10.1534/genetics.115.183665
RAD Capture (Rapture): Flexible and Efficient Sequence-Based Genotyping
Abstract
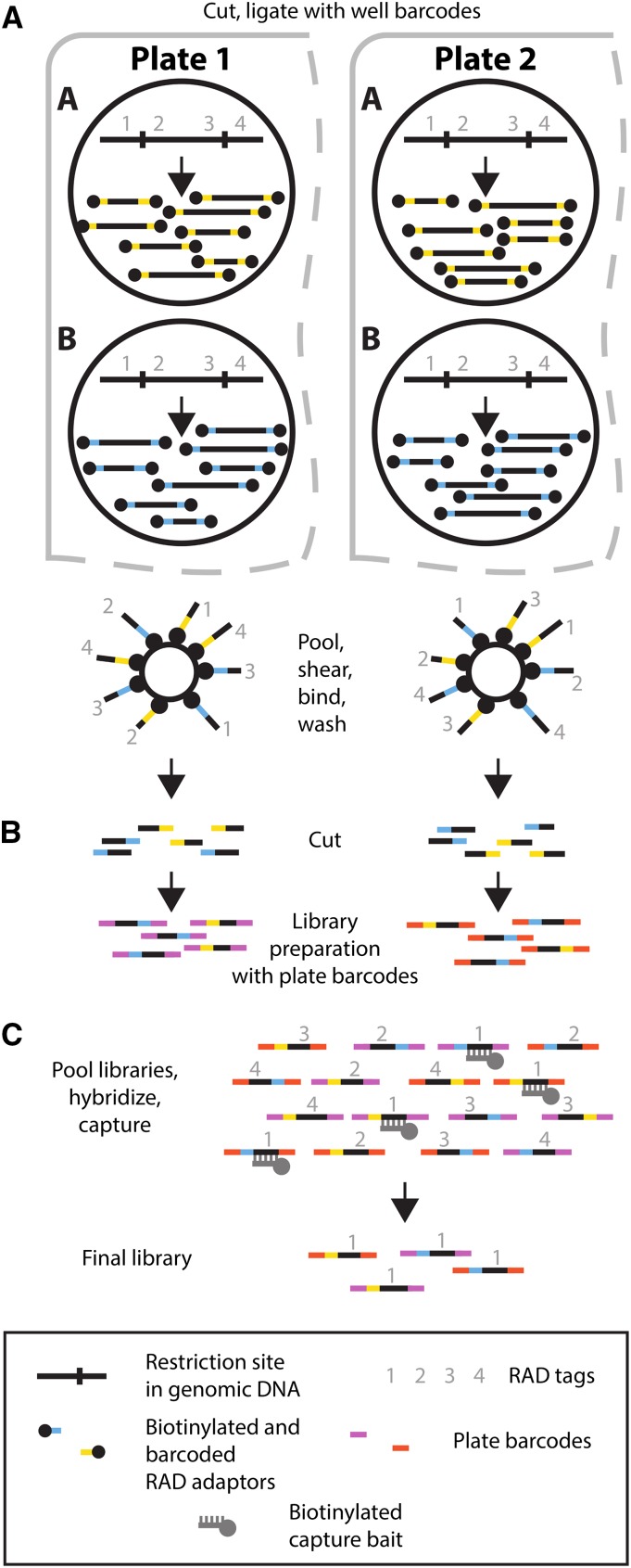
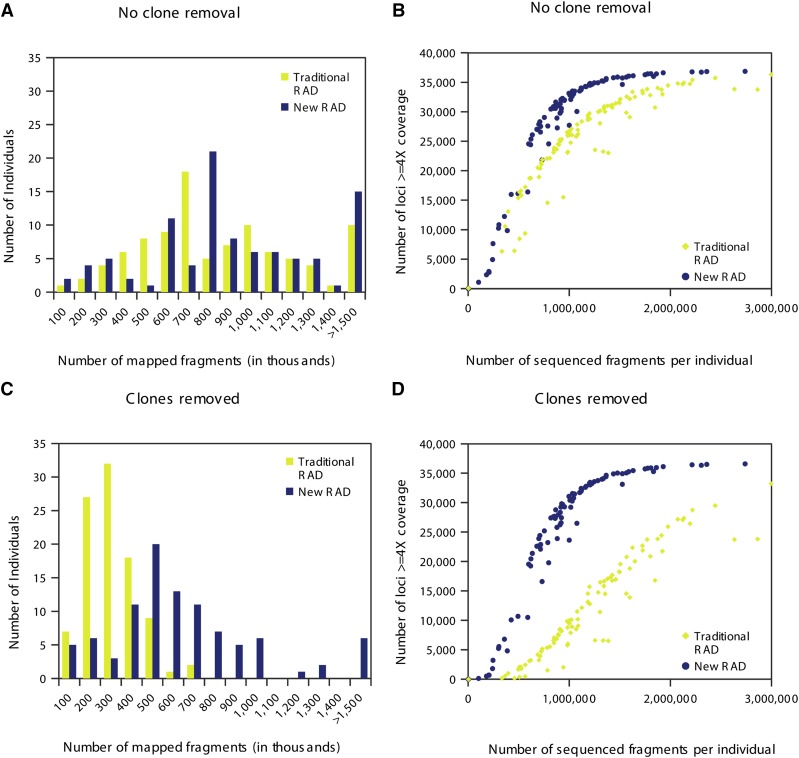
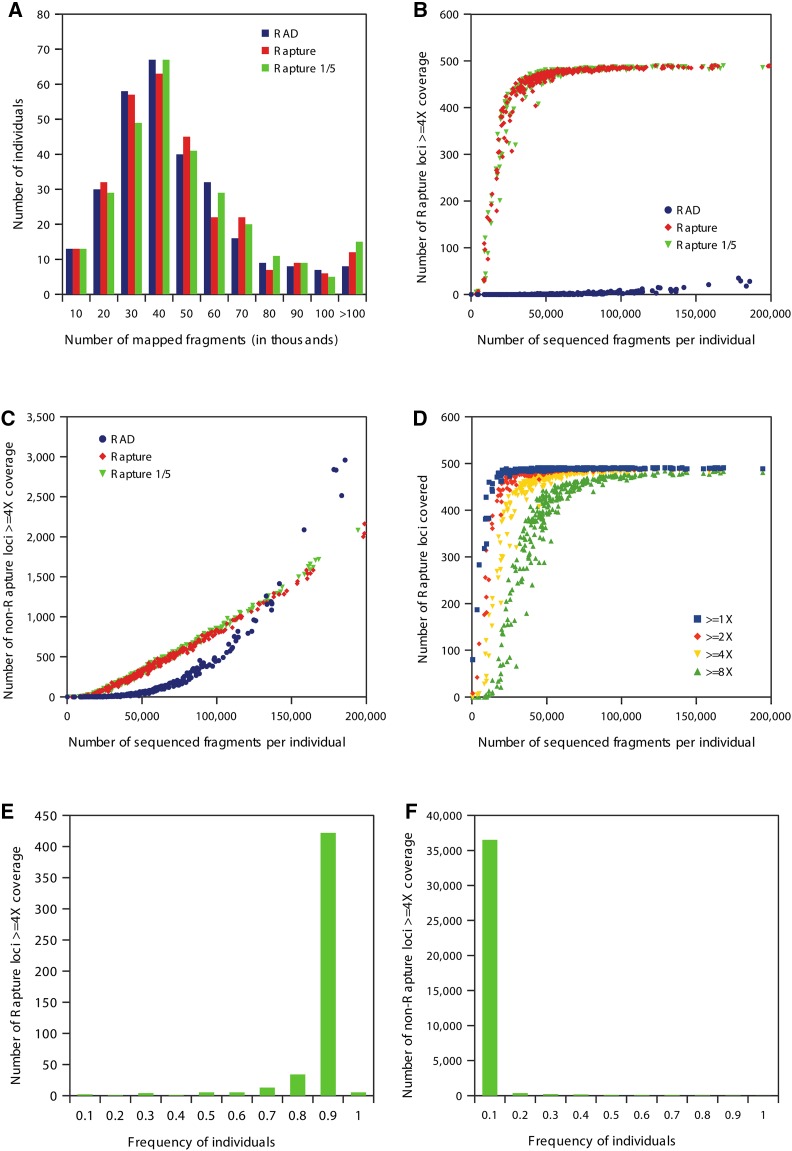
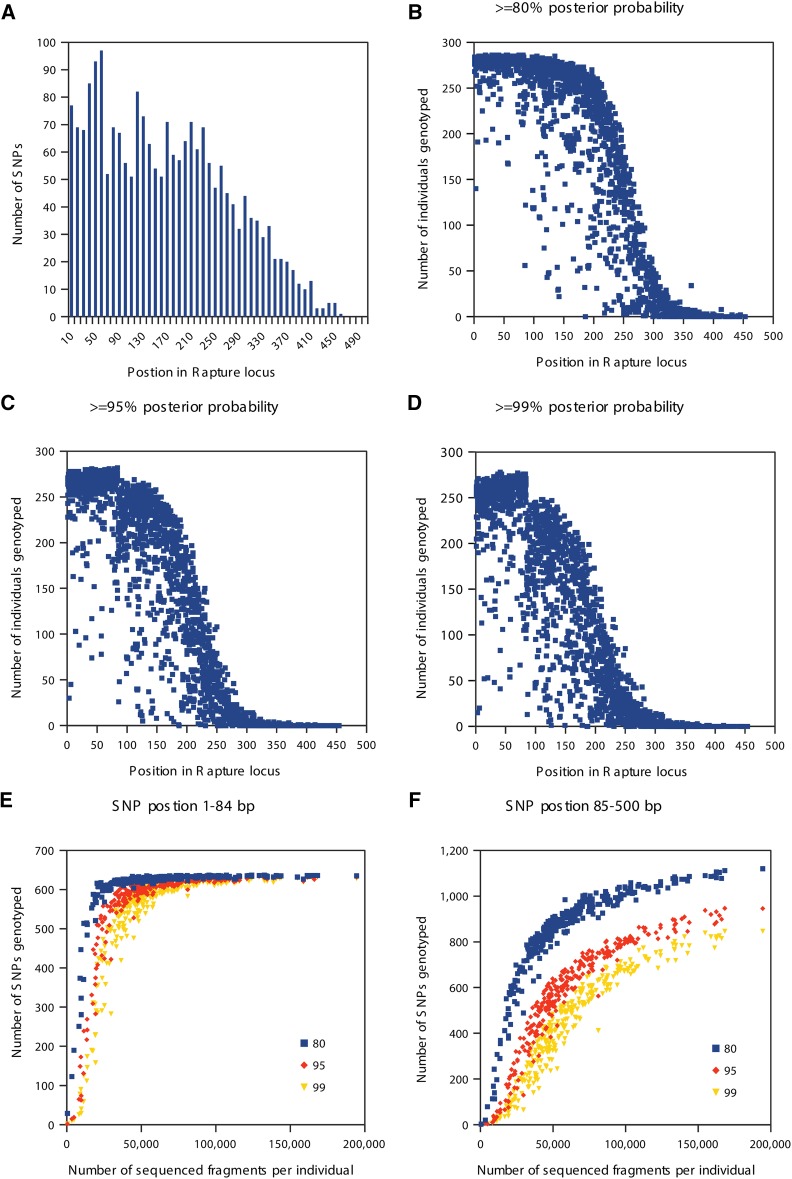
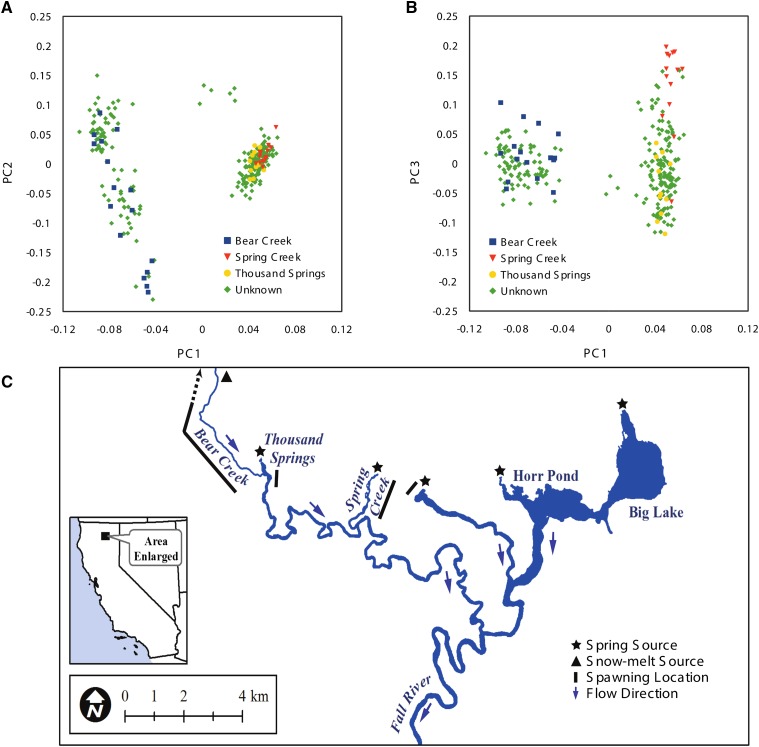
Massively parallel sequencing has revolutionized many areas of biology, but sequencing large amounts of DNA in many individuals is cost-prohibitive and unnecessary for many studies. Genomic complexity reduction techniques such as sequence capture and restriction enzyme-based methods enable the analysis of many more individuals per unit cost. Despite their utility, current complexity reduction methods have limitations, especially when large numbers of individuals are analyzed. Here we develop a much improved restriction site-associated DNA (RAD) sequencing protocol and a new method called Rapture ( R: AD c APTURE: ). The new RAD protocol improves versatility by separating RAD tag isolation and sequencing library preparation into two distinct steps. This protocol also recovers more unique (nonclonal) RAD fragments, which improves both standard RAD and Rapture analysis. Rapture then uses an in-solution capture of chosen RAD tags to target sequencing reads to desired loci. Rapture combines the benefits of both RAD and sequence capture, i.e., very inexpensive and rapid library preparation for many individuals as well as high specificity in the number and location of genomic loci analyzed. Our results demonstrate that Rapture is a rapid and flexible technology capable of analyzing a very large number of individuals with minimal sequencing and library preparation cost. The methods presented here should improve the efficiency of genetic analysis for many aspects of agricultural, environmental, and biomedical science.
Keywords: genotyping; massively parallel sequencing; population genetics; rainbow trout; restriction-site associated DNA (RAD); sequence capture.
Copyright © 2016 by the Genetics Society of America.
Figures
References
-
- Andrews K. R., Hohenlohe P. A., Miller M. R., Hand B. K., Seeb J. E., et al. , 2014. Trade‐offs and utility of alternative RADseq methods: reply to Puritz et al. Mol. Ecol. 23: 5943–5946. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
