

Epidemic Reconstruction in a Phylogenetics Framework: Transmission Trees as Partitions of the Node Set
- PMID: 26717515
- PMCID: PMC4701012
- DOI: 10.1371/journal.pcbi.1004613
Epidemic Reconstruction in a Phylogenetics Framework: Transmission Trees as Partitions of the Node Set
Abstract
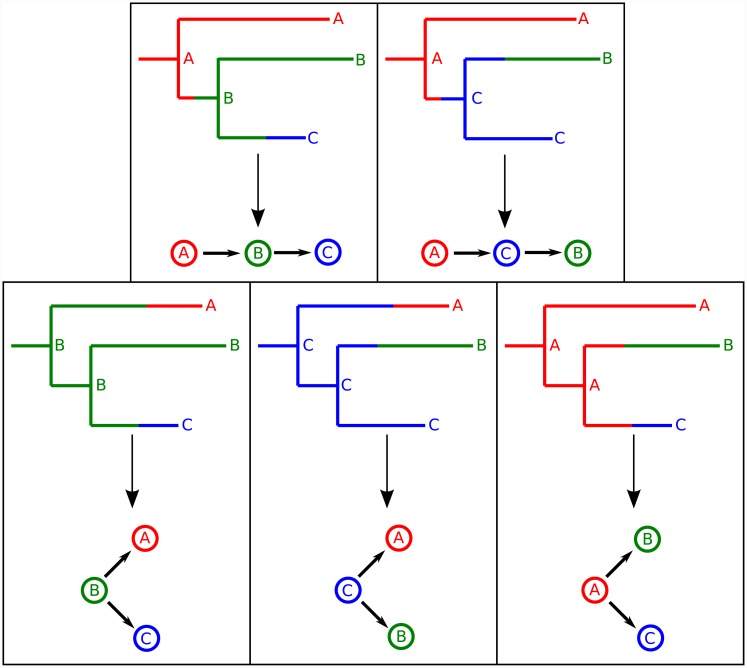
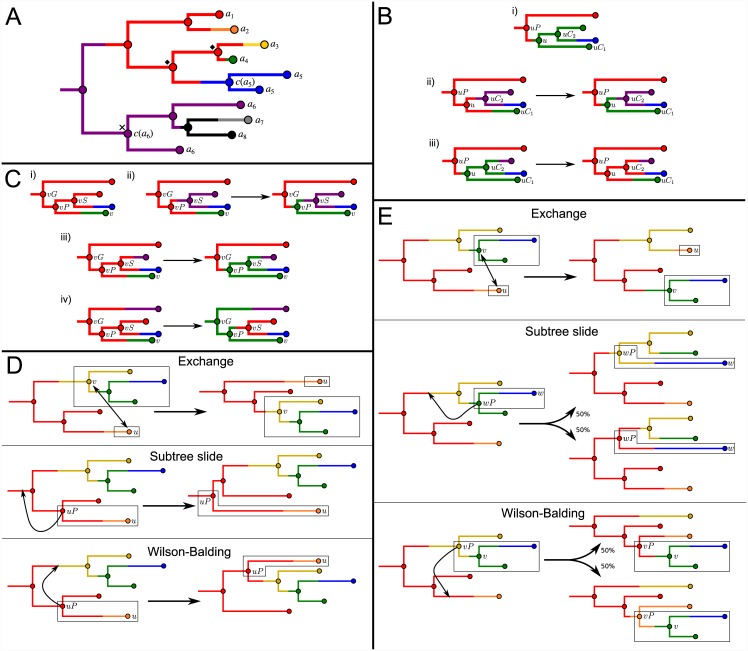
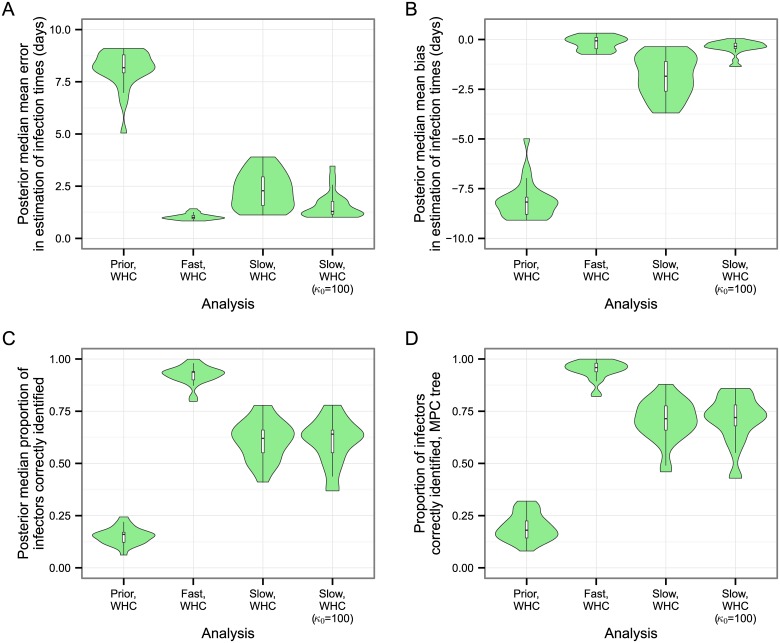
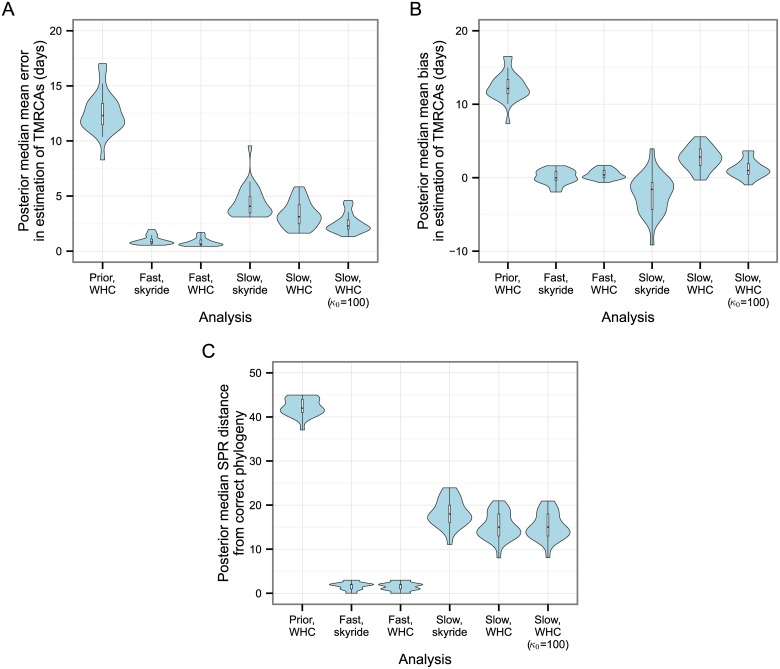
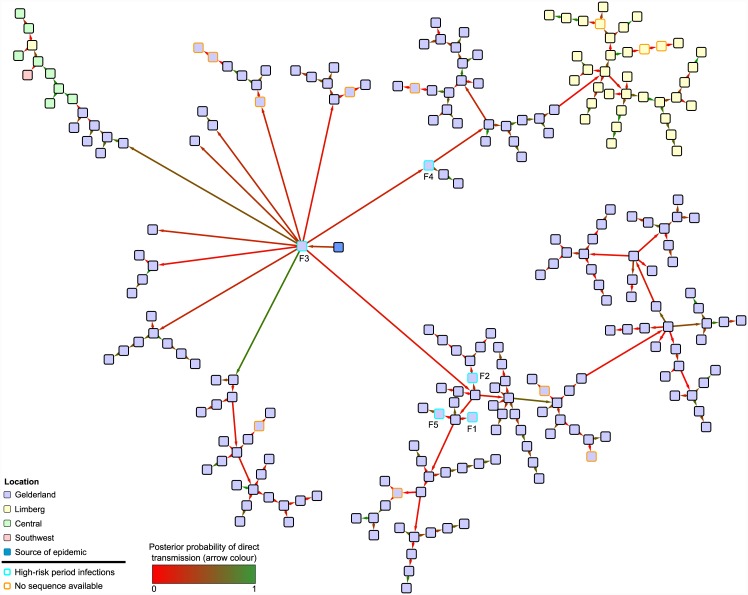
The use of genetic data to reconstruct the transmission tree of infectious disease epidemics and outbreaks has been the subject of an increasing number of studies, but previous approaches have usually either made assumptions that are not fully compatible with phylogenetic inference, or, where they have based inference on a phylogeny, have employed a procedure that requires this tree to be fixed. At the same time, the coalescent-based models of the pathogen population that are employed in the methods usually used for time-resolved phylogeny reconstruction are a considerable simplification of epidemic process, as they assume that pathogen lineages mix freely. Here, we contribute a new method that is simultaneously a phylogeny reconstruction method for isolates taken from an epidemic, and a procedure for transmission tree reconstruction. We observe that, if one or more samples is taken from each host in an epidemic or outbreak and these are used to build a phylogeny, a transmission tree is equivalent to a partition of the set of nodes of this phylogeny, such that each partition element is a set of nodes that is connected in the full tree and contains all the tips corresponding to samples taken from one and only one host. We then implement a Monte Carlo Markov Chain (MCMC) procedure for simultaneous sampling from the spaces of both trees, utilising a newly-designed set of phylogenetic tree proposals that also respect node partitions. We calculate the posterior probability of these partitioned trees based on a model that acknowledges the population structure of an epidemic by employing an individual-based disease transmission model and a coalescent process taking place within each host. We demonstrate our method, first using simulated data, and then with sequences taken from the H7N7 avian influenza outbreak that occurred in the Netherlands in 2003. We show that it is superior to established coalescent methods for reconstructing the topology and node heights of the phylogeny and performs well for transmission tree reconstruction when the phylogeny is well-resolved by the genetic data, but caution that this will often not be the case in practice and that existing genetic and epidemiological data should be used to configure such analyses whenever possible. This method is available for use by the research community as part of BEAST, one of the most widely-used packages for reconstruction of dated phylogenies.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Spada E, Sagliocca L, Sourdis J, Garbuglia AR, Poggi V, Fusco CD, et al. Use of the minimum spanning tree model for molecular epidemiological investigation of a nosocomial outbreak of hepatitis C virus infection. J Clin Microbiol. 2004;42:4230–4236. 10.1128/JCM.42.9.4230-4236.2004 - DOI - PMC - PubMed
-
- Aldrin M, Lyngstad TM, Kristoffersen AB, Storvik B, Borgan Ø, Jansen PA. Modelling the spread of infectious salmon anaemia among salmon farms based on seaway distances between farms and genetic relationships between infectious salmon anaemia virus isolates. J Roy Soc Interface. 2011;8:1346–1356. 10.1098/rsif.2010.0737 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
