

Complement Component 3 Is Regulated by TWIST1 and Mediates Epithelial-Mesenchymal Transition
- PMID: 26718342
- PMCID: PMC4724537
- DOI: 10.4049/jimmunol.1501886
Complement Component 3 Is Regulated by TWIST1 and Mediates Epithelial-Mesenchymal Transition
Abstract
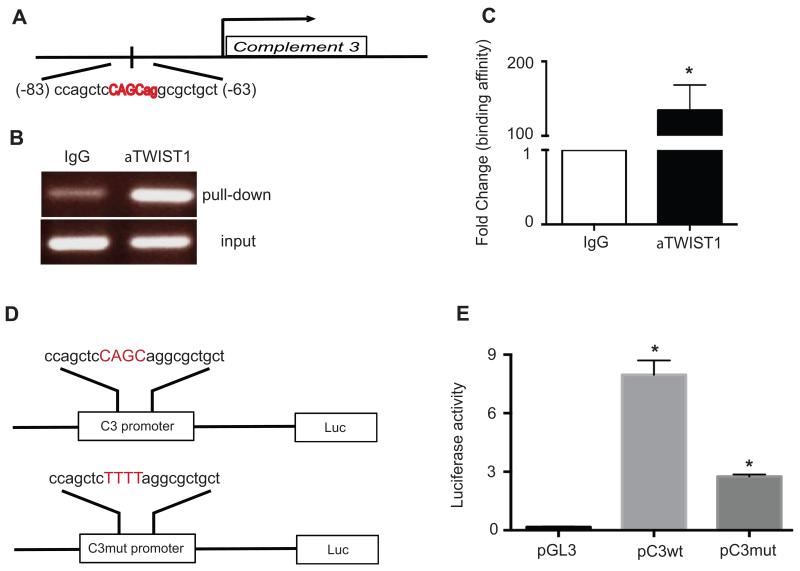
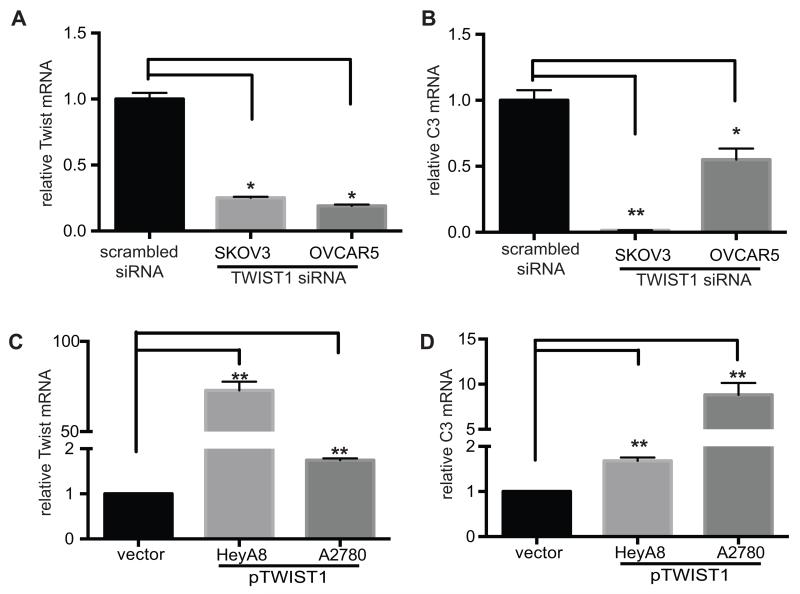
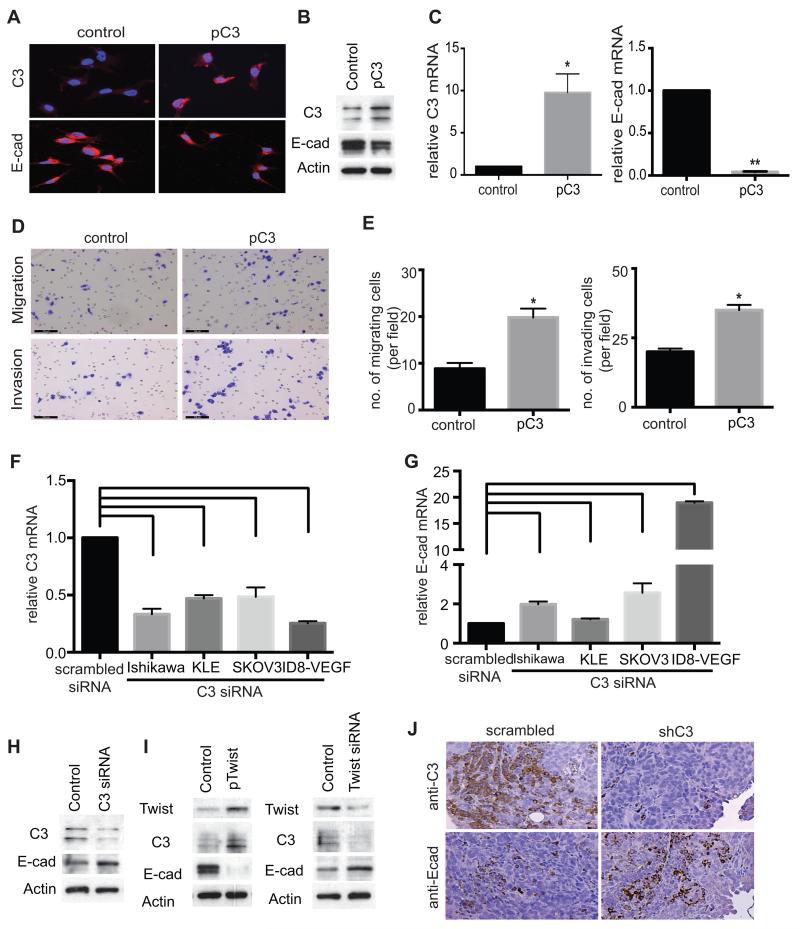
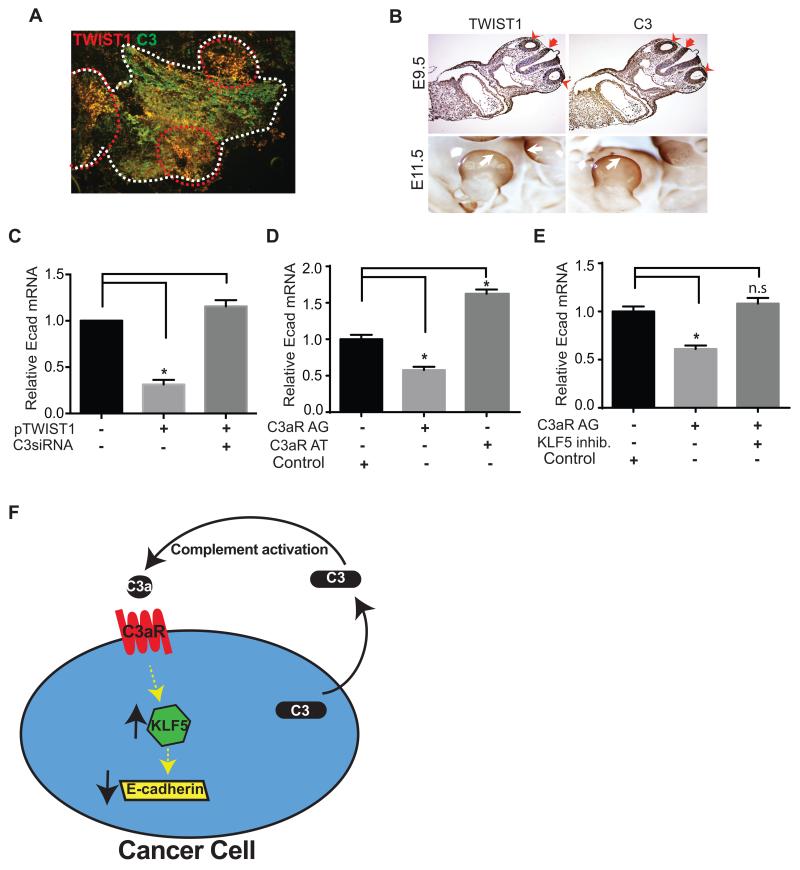
We have previously shown that complement component 3 (C3) is secreted by malignant epithelial cells. To understand the mechanism of upregulation of C3 expression in tumor cells, we studied the C3 promoter and identified that twist basic helix-loop-helix transcription factor 1 (TWIST1) binds to the C3 promoter and enhances its expression. Because TWIST1 mediates epithelial-mesenchymal transition (EMT), we studied the effect of C3 on EMT and found that C3 decreased E-cadherin expression on cancer cells and promoted EMT. We showed that C3-induced reduction in E-cadherin expression in ovarian cancer cells was mediated by C3a and is Krüppel-like factor 5 dependent. We investigated the association between TWIST1 and C3 in malignant tumors and in murine embryos. TWIST1 and C3 colocalized at the invasive tumor edges, and in the neural crest and limb buds of mouse embryos. Our results identified TWIST1 as a transcription factor that regulates C3 expression during pathologic and physiologic EMT.
Copyright © 2016 by The American Association of Immunologists, Inc.
Figures
References
-
- Mastellos D, Lambris JD. Complement: more than a ‘guard’ against invading pathogens? Trends Immunol. 2002;23:485–491. - PubMed
-
- McLin VA, Hu CH, Shah R, Jamrich M. Expression of complement components coincides with early patterning and organogenesis in Xenopus laevis. Int. J. Dev. Biol. 2008;52:1123–1133. - PubMed
-
- Raedler H, Yang M, Lalli PN, Medof ME, Heeger PS. Primed CD8(+) T-cell responses to allogeneic endothelial cells are controlled by local complement activation. Am. J. Transplant. 2009;9:1784–1795. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
