

Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing
- PMID: 26718401
- PMCID: PMC4830092
- DOI: 10.1016/j.bbrc.2015.12.083
Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing
Abstract
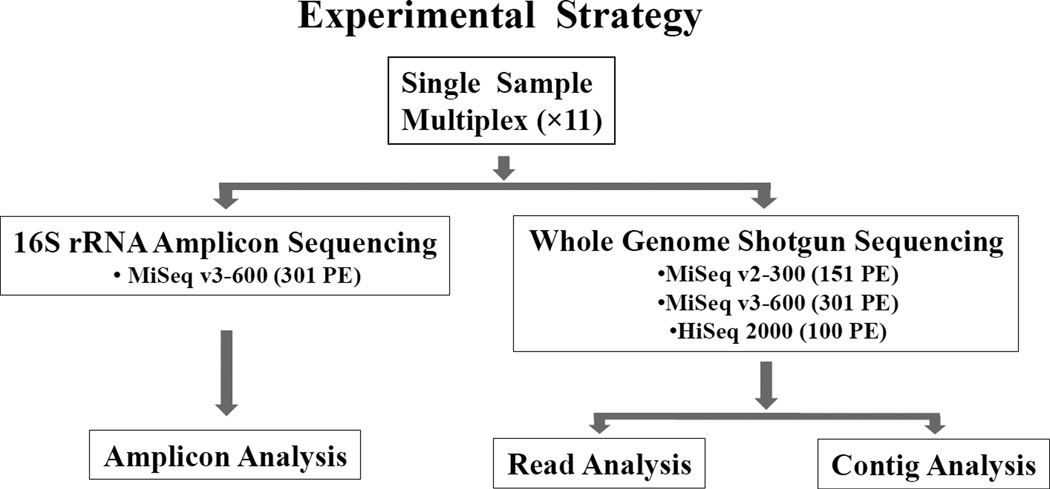
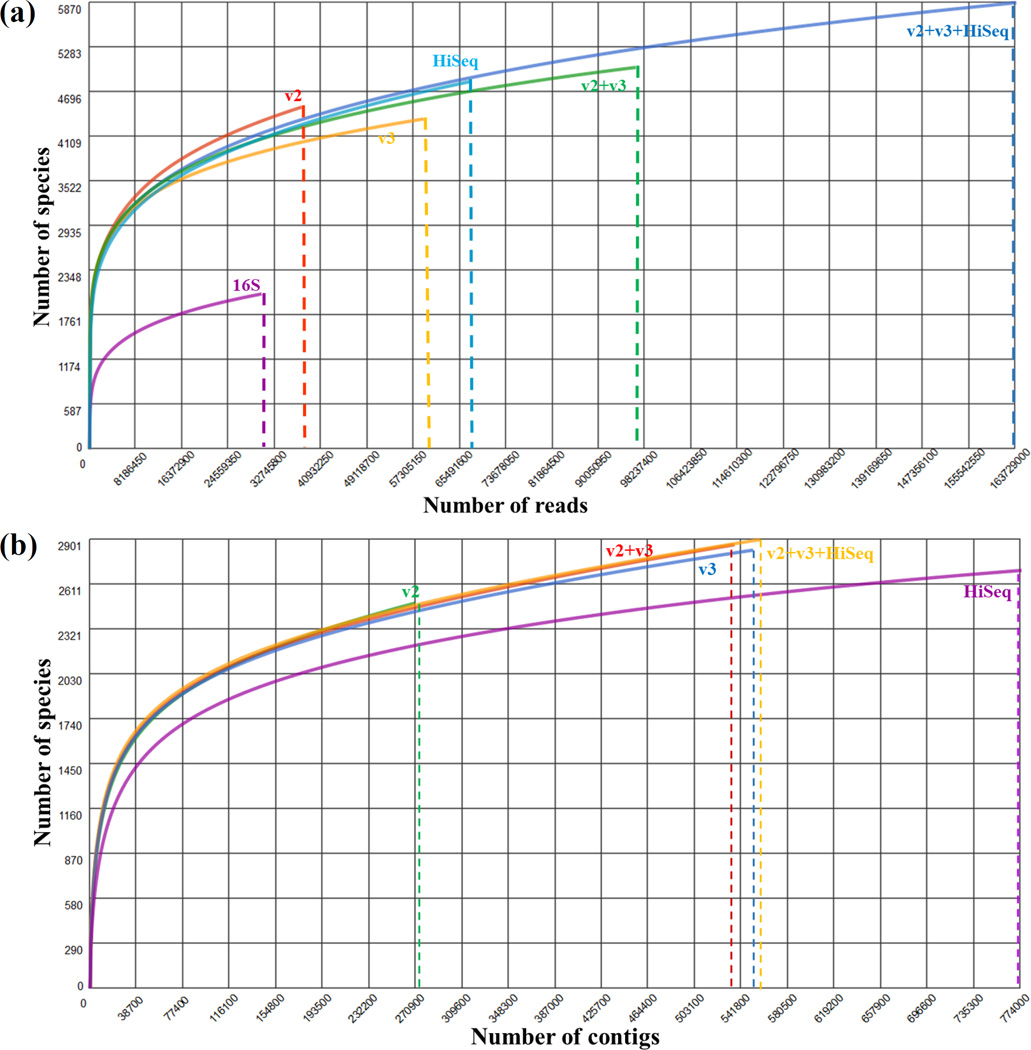
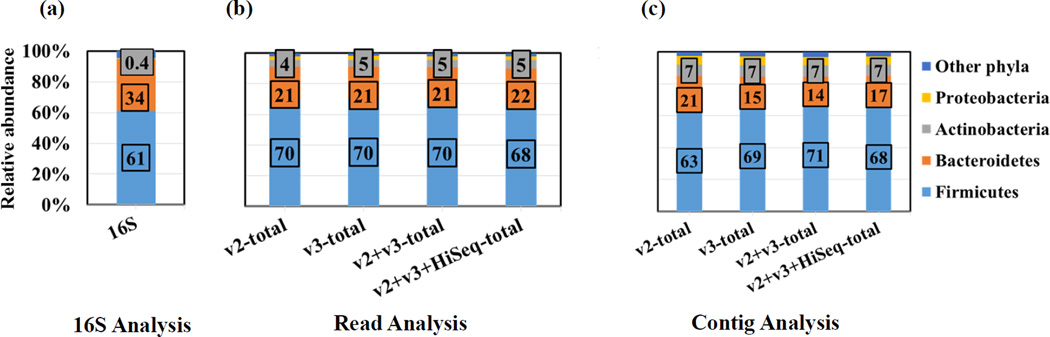
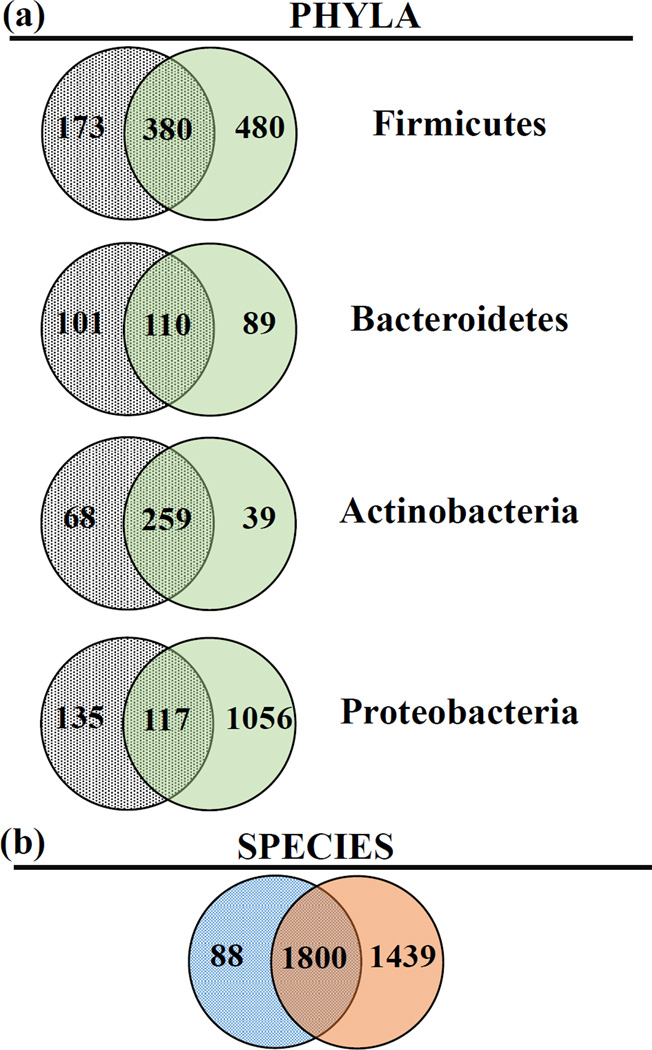
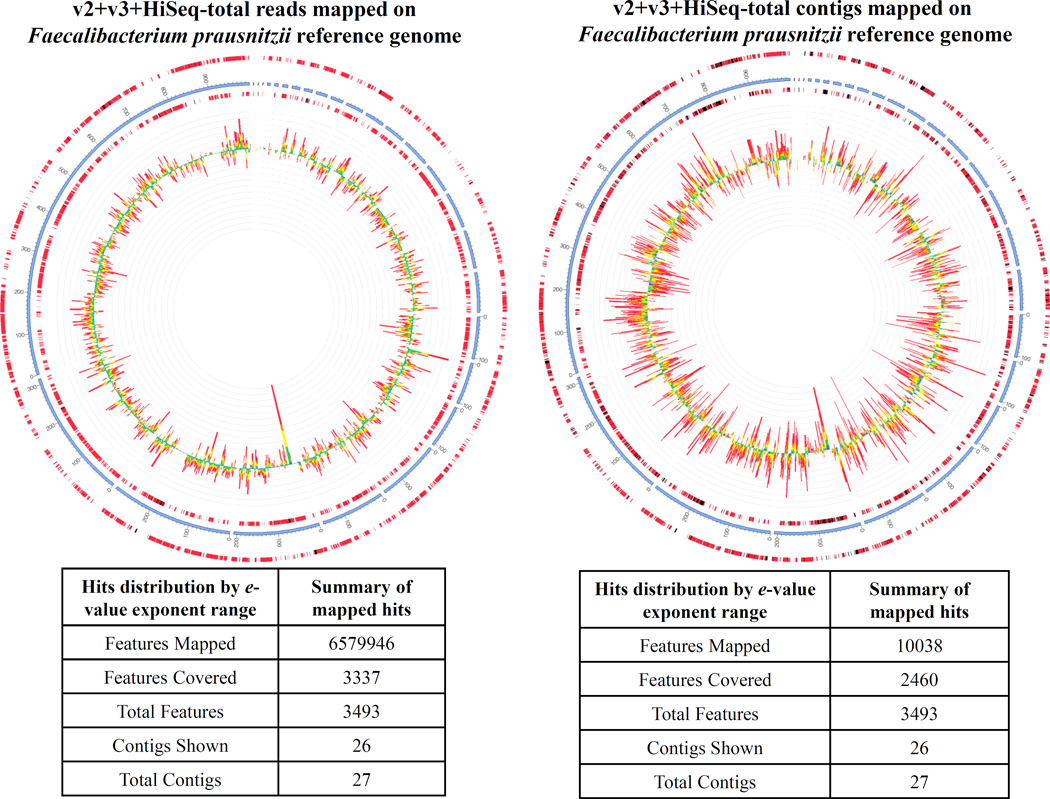
The human microbiome has emerged as a major player in regulating human health and disease. Translational studies of the microbiome have the potential to indicate clinical applications such as fecal transplants and probiotics. However, one major issue is accurate identification of microbes constituting the microbiota. Studies of the microbiome have frequently utilized sequencing of the conserved 16S ribosomal RNA (rRNA) gene. We present a comparative study of an alternative approach using whole genome shotgun sequencing (WGS). In the present study, we analyzed the human fecal microbiome compiling a total of 194.1 × 10(6) reads from a single sample using multiple sequencing methods and platforms. Specifically, after establishing the reproducibility of our methods with extensive multiplexing, we compared: 1) The 16S rRNA amplicon versus the WGS method, 2) the Illumina HiSeq versus MiSeq platforms, 3) the analysis of reads versus de novo assembled contigs, and 4) the effect of shorter versus longer reads. Our study demonstrates that whole genome shotgun sequencing has multiple advantages compared with the 16S amplicon method including enhanced detection of bacterial species, increased detection of diversity and increased prediction of genes. In addition, increased length, either due to longer reads or the assembly of contigs, improved the accuracy of species detection.
Keywords: 16S rRNA; Amplicon sequencing; Metagenomics; Microbiome; Microbiota; Next-generation sequencing; Whole genome shotgun sequencing.
Copyright © 2015 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors have declared that no conflict of interest exists.
Figures

References
-
- Pflughoeft KJ, Versalovic J. Human microbiome in health and disease. Annu. Rev. Pathol. 2012;7:99–122. - PubMed
-
- Kelly CP. Fecal microbiota transplantation — An old therapy comes of age. N. Engl. J. Med. 2013;368:474–475. - PubMed
-
- Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, Derrien M, Druesne A, Van Hylckama Vlieg JE, Bloks VW, Groen AK, Heilig HG, Zoetendal EG, Stroes ES, de Vos WM, Hoekstra JB, Nieuwdorp M. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143:913–916. e917. - PubMed
-
- Ratner M. Fecal transplantation poses dilemma for FDA. Nat. Biotechnol. 2014;32:401–402. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
