

Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome
- PMID: 26719772
- PMCID: PMC4696349
- DOI: 10.1186/s13072-015-0050-4
Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome
Abstract
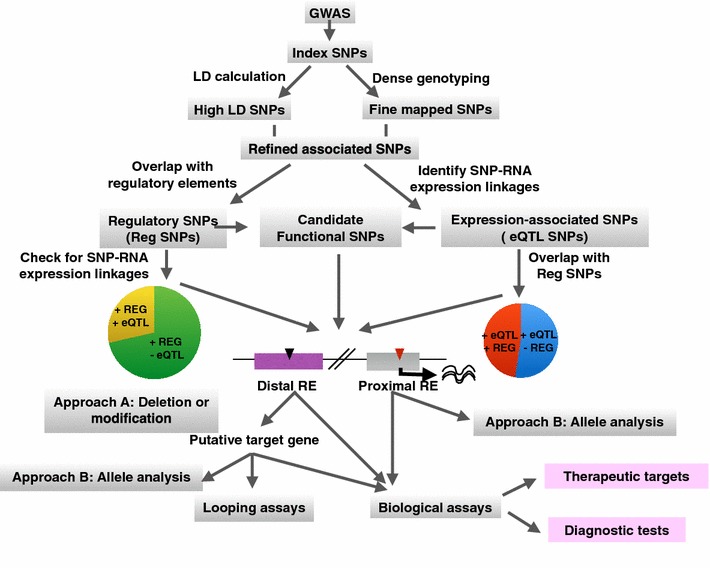
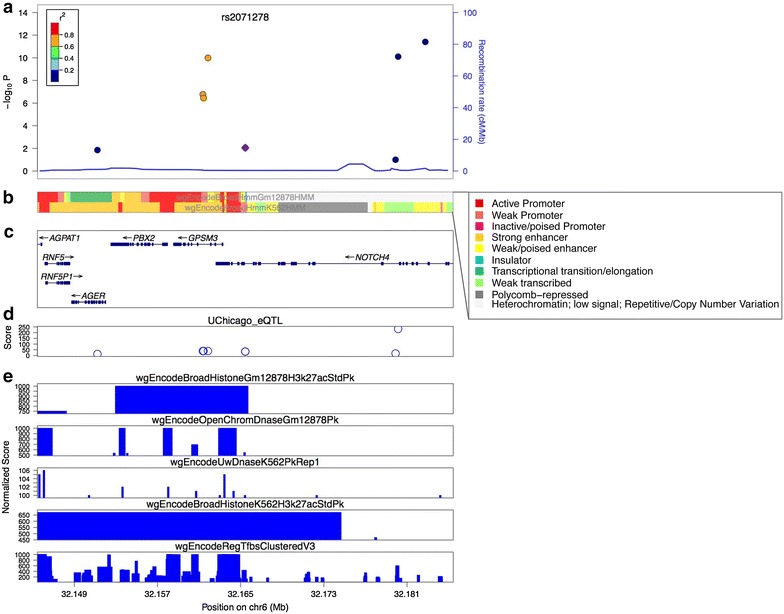
Considerable progress towards an understanding of complex diseases has been made in recent years due to the development of high-throughput genotyping technologies. Using microarrays that contain millions of single-nucleotide polymorphisms (SNPs), Genome Wide Association Studies (GWASs) have identified SNPs that are associated with many complex diseases or traits. For example, as of February 2015, 2111 association studies have identified 15,396 SNPs for various diseases and traits, with the number of identified SNP-disease/trait associations increasing rapidly in recent years. However, it has been difficult for researchers to understand disease risk from GWAS results. This is because most GWAS-identified SNPs are located in non-coding regions of the genome. It is important to consider that the GWAS-identified SNPs serve only as representatives for all SNPs in the same haplotype block, and it is equally likely that other SNPs in high linkage disequilibrium (LD) with the array-identified SNPs are causal for the disease. Because it was hoped that disease-associated coding variants would be identified if the true casual SNPs were known, investigators have expanded their analyses using LD calculation and fine-mapping. However, such analyses also identified risk-associated SNPs located in non-coding regions. Thus, the GWAS field has been left with the conundrum as to how a single-nucleotide change in a non-coding region could confer increased risk for a specific disease. One possible answer to this puzzle is that the variant SNPs cause changes in gene expression levels rather than causing changes in protein function. This review provides a description of (1) advances in genomic and epigenomic approaches that incorporate functional annotation of regulatory elements to prioritize the disease risk-associated SNPs that are located in non-coding regions of the genome for follow-up studies, (2) various computational tools that aid in identifying gene expression changes caused by the non-coding disease-associated SNPs, and (3) experimental approaches to identify target genes of, and study the biological phenotypes conferred by, non-coding disease-associated SNPs.
Keywords: Enhancers; GWAS; Genome engineering; Non-coding SNPs.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
