

ConBind: motif-aware cross-species alignment for the identification of functional transcription factor binding sites
- PMID: 26721389
- PMCID: PMC4856970
- DOI: 10.1093/nar/gkv1518
ConBind: motif-aware cross-species alignment for the identification of functional transcription factor binding sites
Abstract
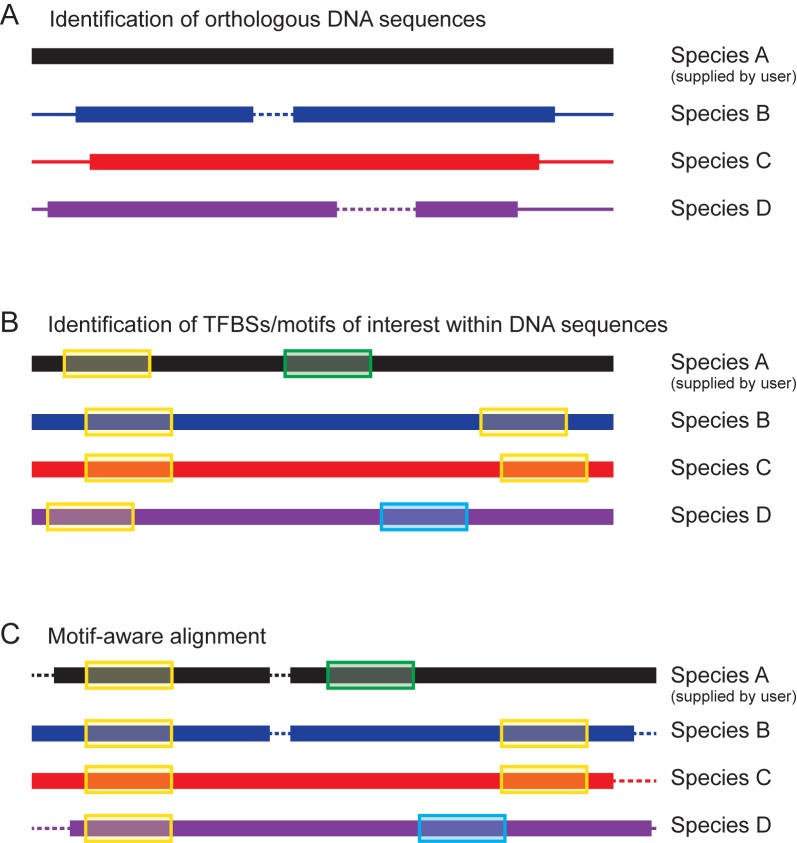
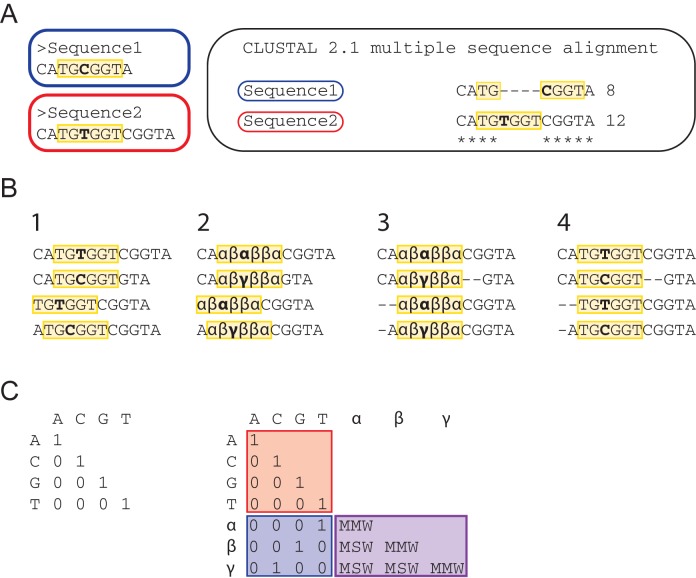
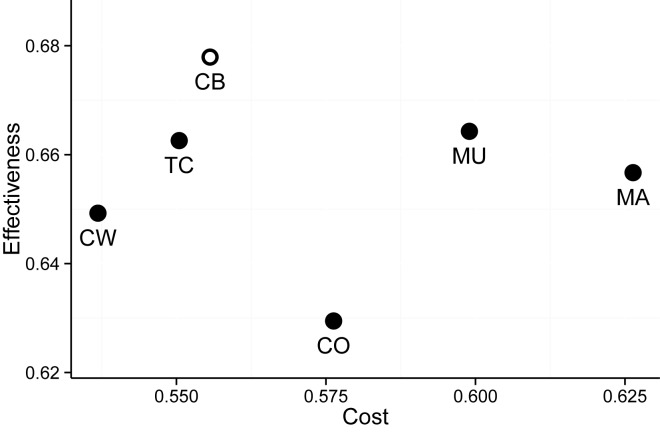
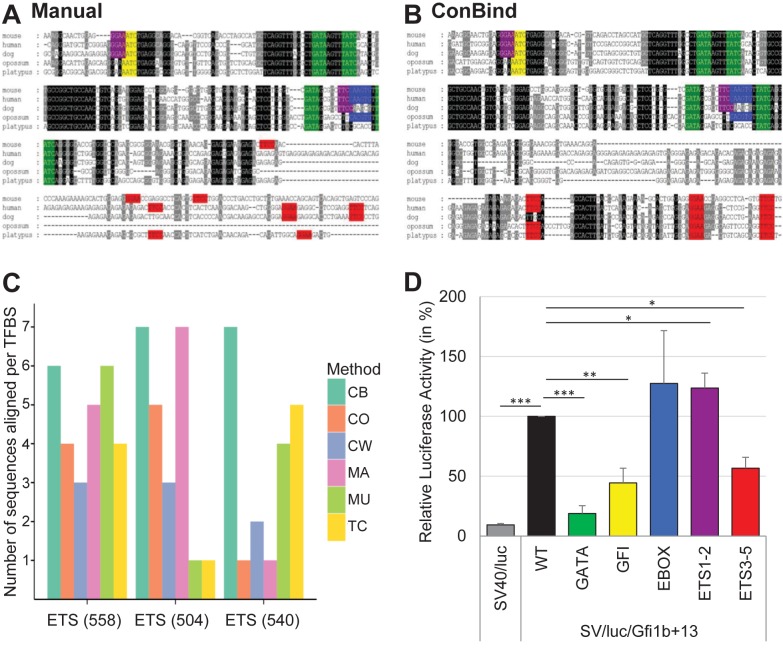
Eukaryotic gene expression is regulated by transcription factors (TFs) binding to promoter as well as distal enhancers. TFs recognize short, but specific binding sites (TFBSs) that are located within the promoter and enhancer regions. Functionally relevant TFBSs are often highly conserved during evolution leaving a strong phylogenetic signal. While multiple sequence alignment (MSA) is a potent tool to detect the phylogenetic signal, the current MSA implementations are optimized to align the maximum number of identical nucleotides. This approach might result in the omission of conserved motifs that contain interchangeable nucleotides such as the ETS motif (IUPAC code: GGAW). Here, we introduce ConBind, a novel method to enhance alignment of short motifs, even if their mutual sequence similarity is only partial. ConBind improves the identification of conserved TFBSs by improving the alignment accuracy of TFBS families within orthologous DNA sequences. Functional validation of the Gfi1b + 13 enhancer reveals that ConBind identifies additional functionally important ETS binding sites that were missed by all other tested alignment tools. In addition to the analysis of known regulatory regions, our web tool is useful for the analysis of TFBSs on so far unknown DNA regions identified through ChIP-sequencing.
© The Author(s) 2015. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Chapman M.A., Charchar F.J., Kinston S., Bird C.P., Grafham D., Rogers J., Grutzner F., Graves J.A., Green A.R., Gottgens B. Comparative and functional analyses of LYL1 loci establish marsupial sequences as a model for phylogenetic footprinting. Genomics. 2003;81:249–259. - PubMed
-
- Donaldson I.J., Chapman M., Kinston S., Landry J.R., Knezevic K., Piltz S., Buckley N., Green A.R., Gottgens B. Genome-wide identification of cis-regulatory sequences controlling blood and endothelial development. Hum. Mol. Genet. 2005;14:595–601. - PubMed
-
- Osawa M., Yamaguchi T., Nakamura Y., Kaneko S., Onodera M., Sawada K.-i., Jegalian A., Wu H., Nakauchi H., Iwama A. Erythroid expansion mediated by the Gfi-1B zinc finger protein: role in normal hematopoiesis. Blood. 2002;100:2769–2777. - PubMed
-
- Vassen L., Okayama T., Moroy T. Gfi1b:green fluorescent protein knock-in mice reveal a dynamic expression pattern of Gfi1b during hematopoiesis that is largely complementary to Gfi1. Blood. 2007;109:2356–2364. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
