

Use of a genetically engineered mouse model as a preclinical tool for HER2 breast cancer
- PMID: 26721874
- PMCID: PMC4770148
- DOI: 10.1242/dmm.023143
Use of a genetically engineered mouse model as a preclinical tool for HER2 breast cancer
Abstract
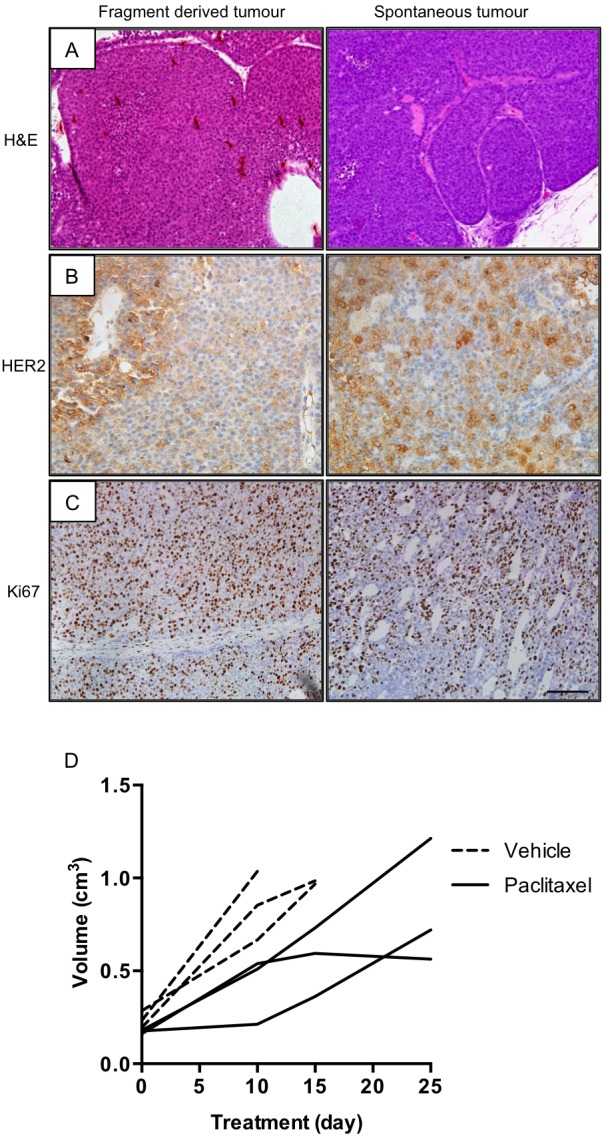
Resistance to human epidermal growth factor receptor 2 (HER2)-targeted therapies presents a major clinical problem. Although preclinical studies have identified a number of possible mechanisms, clinical validation has been difficult. This is most likely to reflect the reliance on cell-line models that do not recapitulate the complexity and heterogeneity seen in human tumours. Here, we show the utility of a genetically engineered mouse model of HER2-driven breast cancer (MMTV-NIC) to define mechanisms of resistance to the pan-HER family inhibitor AZD8931. Genetic manipulation of MMTV-NIC mice demonstrated that loss of phosphatase and tensin homologue (PTEN) conferred de novo resistance to AZD8931, and a tumour fragment transplantation model was established to assess mechanisms of acquired resistance. Using this approach, 50% of tumours developed resistance to AZD8931. Analysis of the resistant tumours showed two distinct patterns of resistance: tumours in which reduced membranous HER2 expression was associated with an epithelial-to-mesenchymal transition (EMT) and resistant tumours that retained HER2 expression and an epithelial morphology. The plasticity of the EMT phenotype was demonstrated upon re-implantation of resistant tumours that then showed a mixed epithelial and mesenchymal phenotype. Further AZD8931 treatment resulted in the generation of secondary resistant tumours that again had either undergone EMT or retained their original epithelial morphology. The data provide a strong rationale for basing therapeutic decisions on the biology of the individual resistant tumour, which can be very different from that of the primary tumour and will be specific to individual patients.
Keywords: Breast cancer; Epithelial-to-mesenchymal transition; HER2; Resistance.
© 2016. Published by The Company of Biologists Ltd.
Conflict of interest statement
T.K. and L.A.B. are employees of AstraZeneca. All other authors have no competing interests.
Figures






References
-
- Berns K., Horlings H. M., Hennessy B. T., Madiredjo M., Hijmans E. M., Beelen K., Linn S. C., Gonzalez-Angulo A. M., Stemke-Hale K., Hauptmann M. et al. (2007). A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 12, 395-402. 10.1016/j.ccr.2007.08.030 - DOI - PubMed
-
- Esteva F. J., Guo H., Zhang S., Santa-Maria C., Stone S., Lanchbury J. S., Sahin A. A., Hortobagyi G. N. and Yu D. (2010). PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am. J. Pathol. 177, 1647-1656. 10.2353/ajpath.2010.090885 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
