

NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism
- PMID: 26724859
- PMCID: PMC4715587
- DOI: 10.1016/j.cmet.2015.12.004
NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism
Erratum in
-
NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism.Cell Metab. 2025 Oct 7;37(10):2093-2095. doi: 10.1016/j.cmet.2025.08.007. Epub 2025 Aug 28. Cell Metab. 2025. PMID: 40876450 Free PMC article. No abstract available.
Abstract
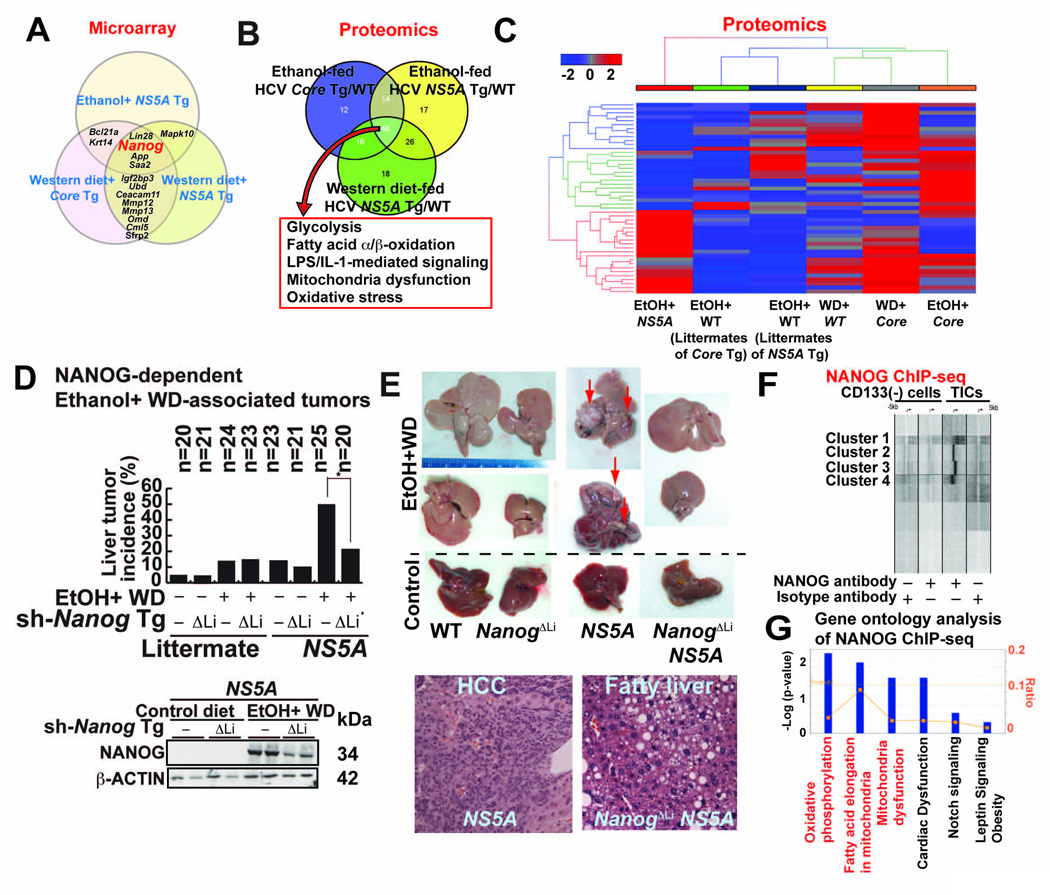
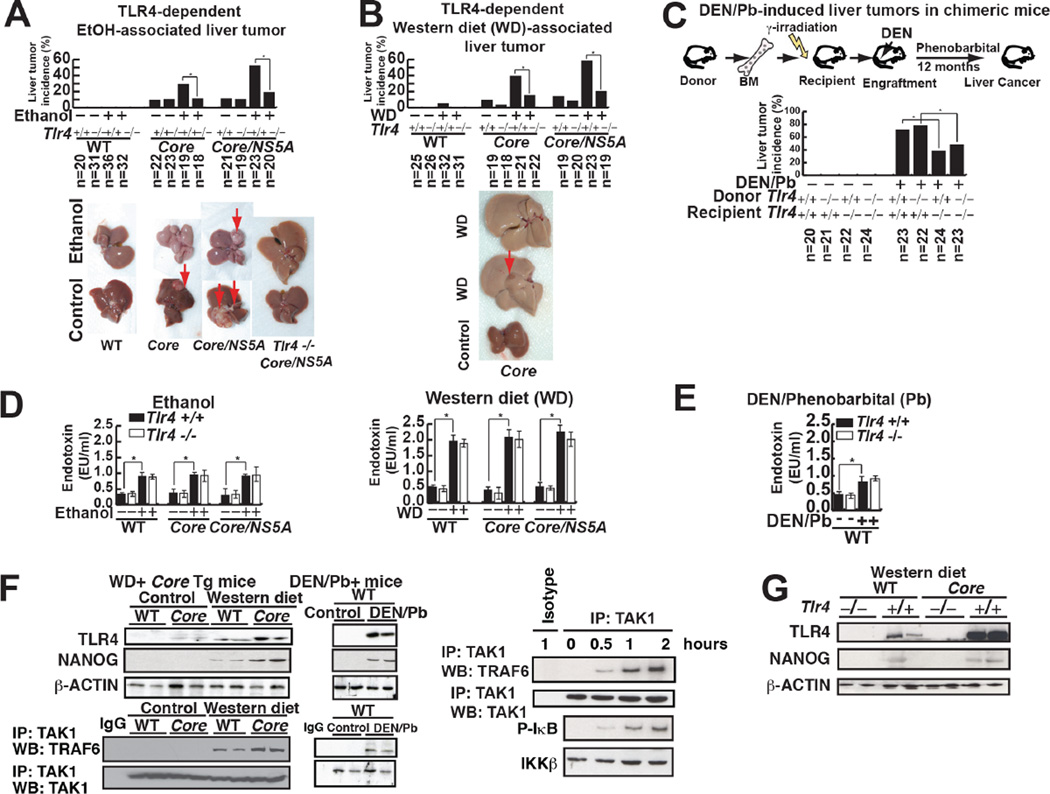
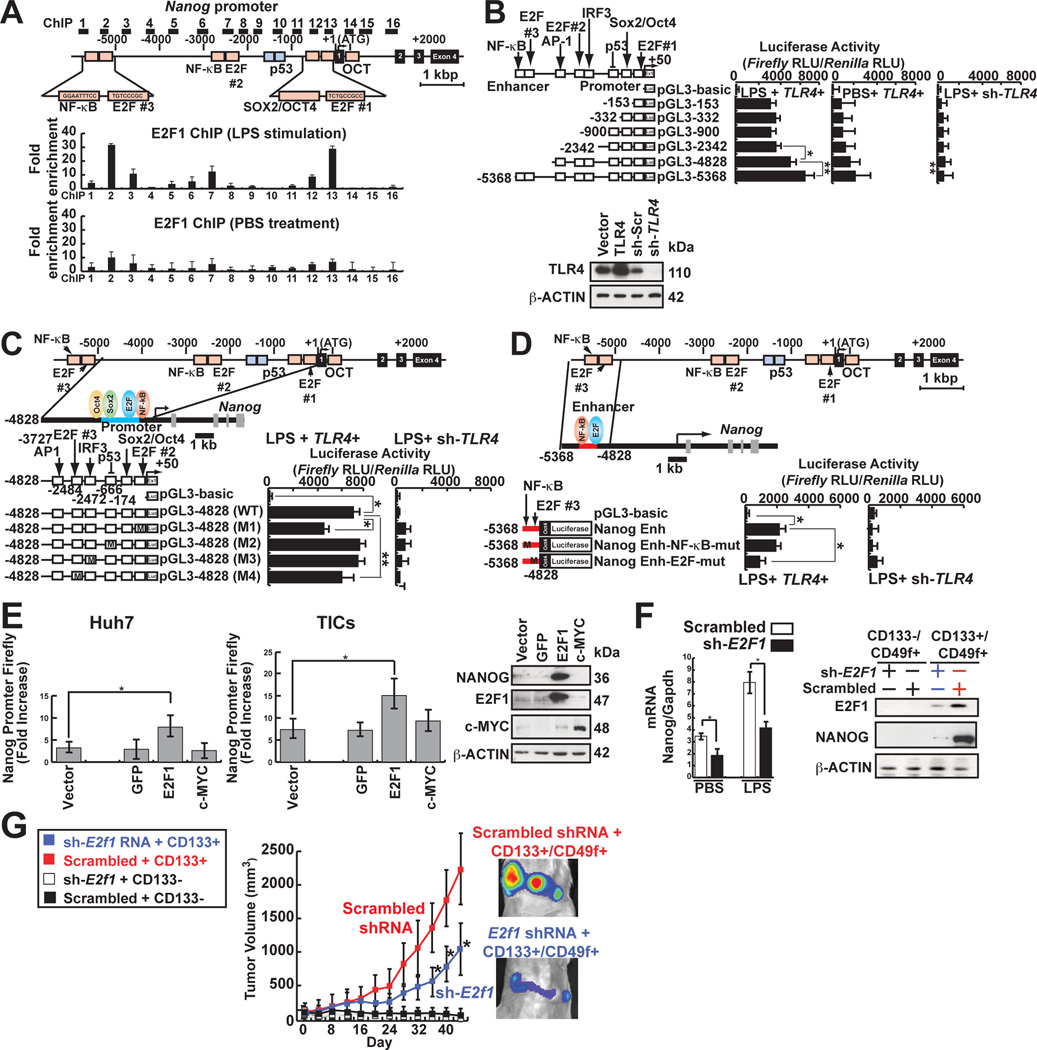
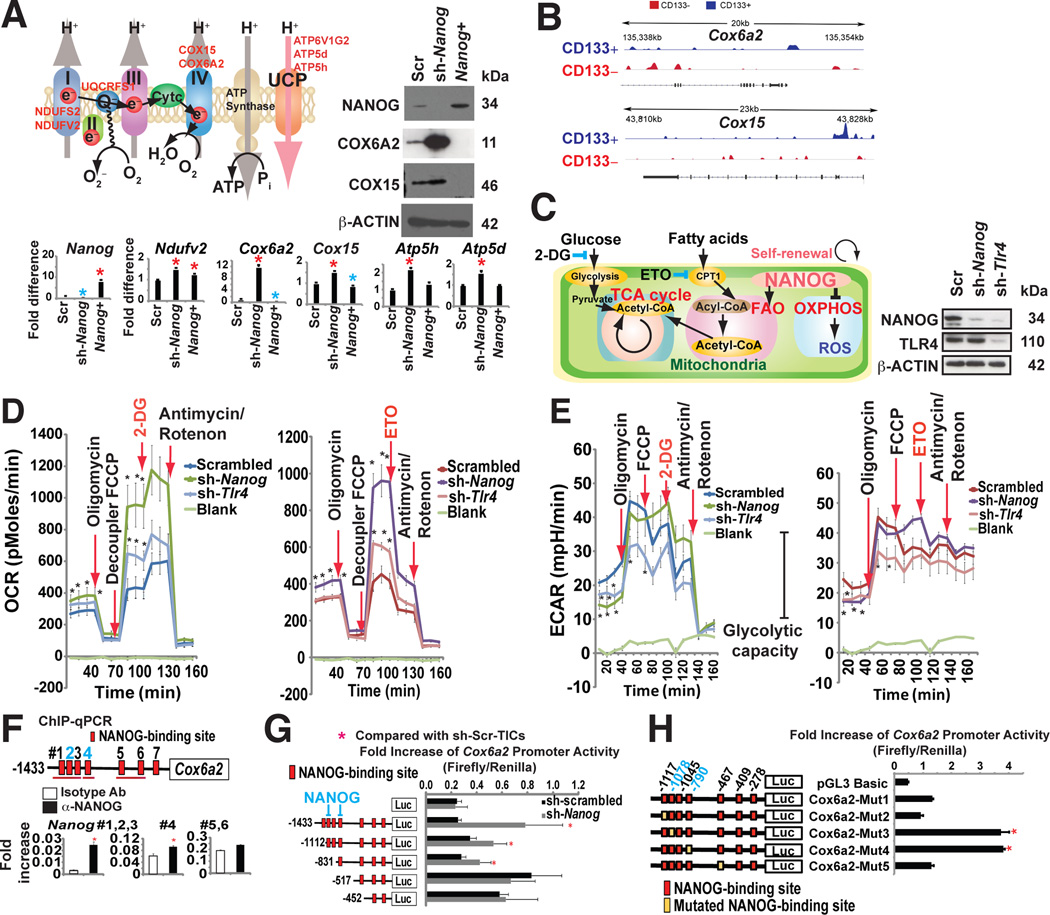
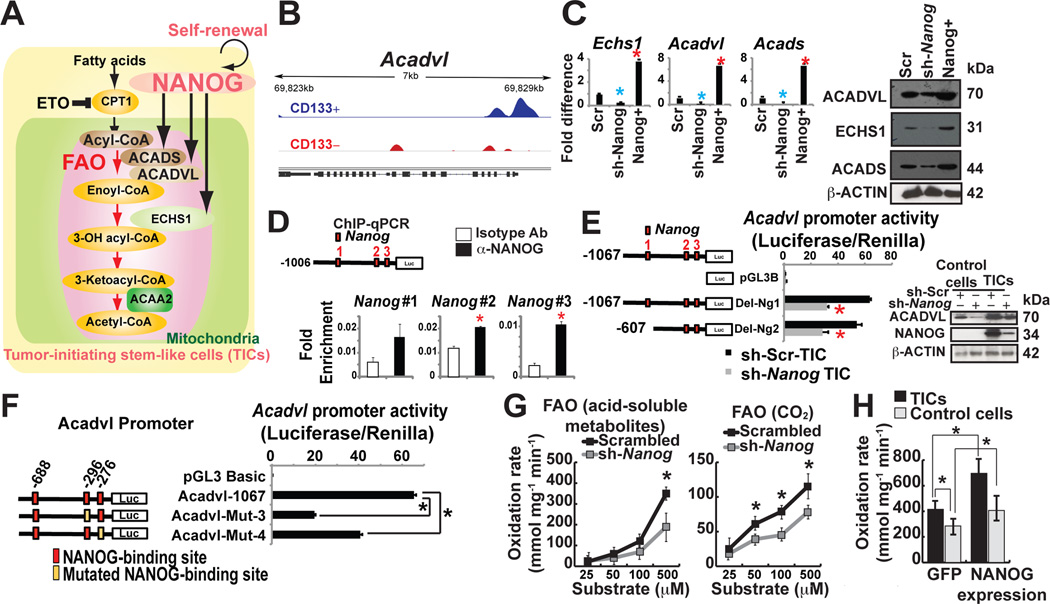
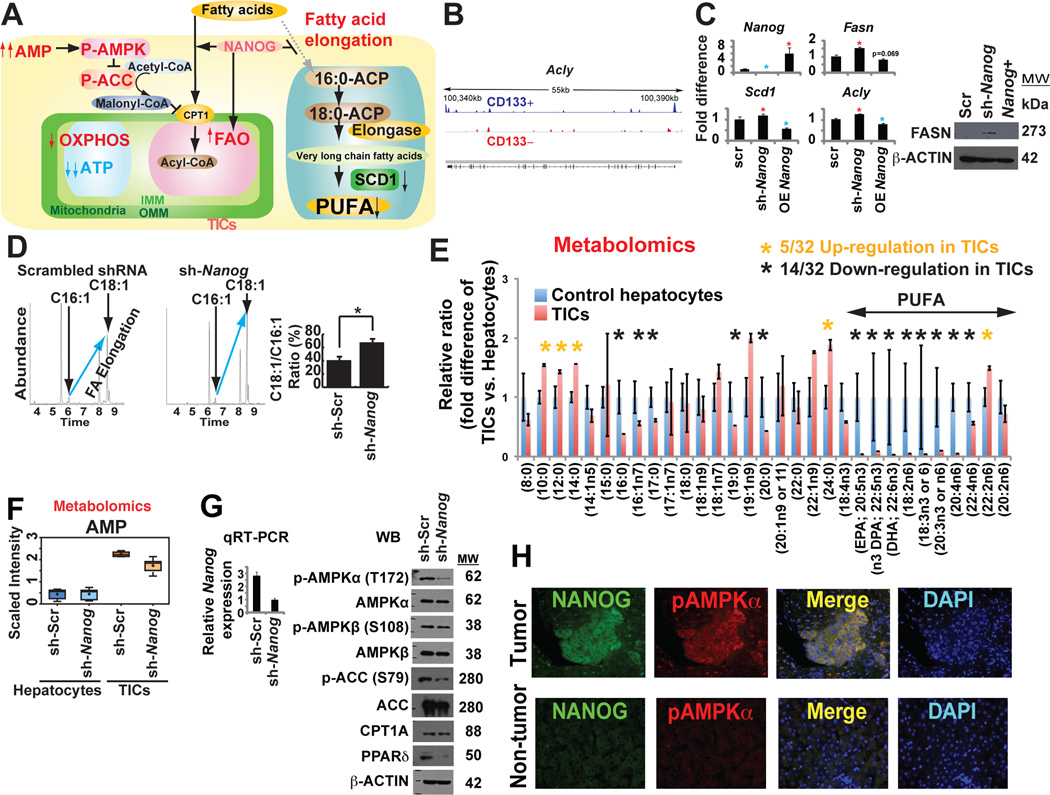
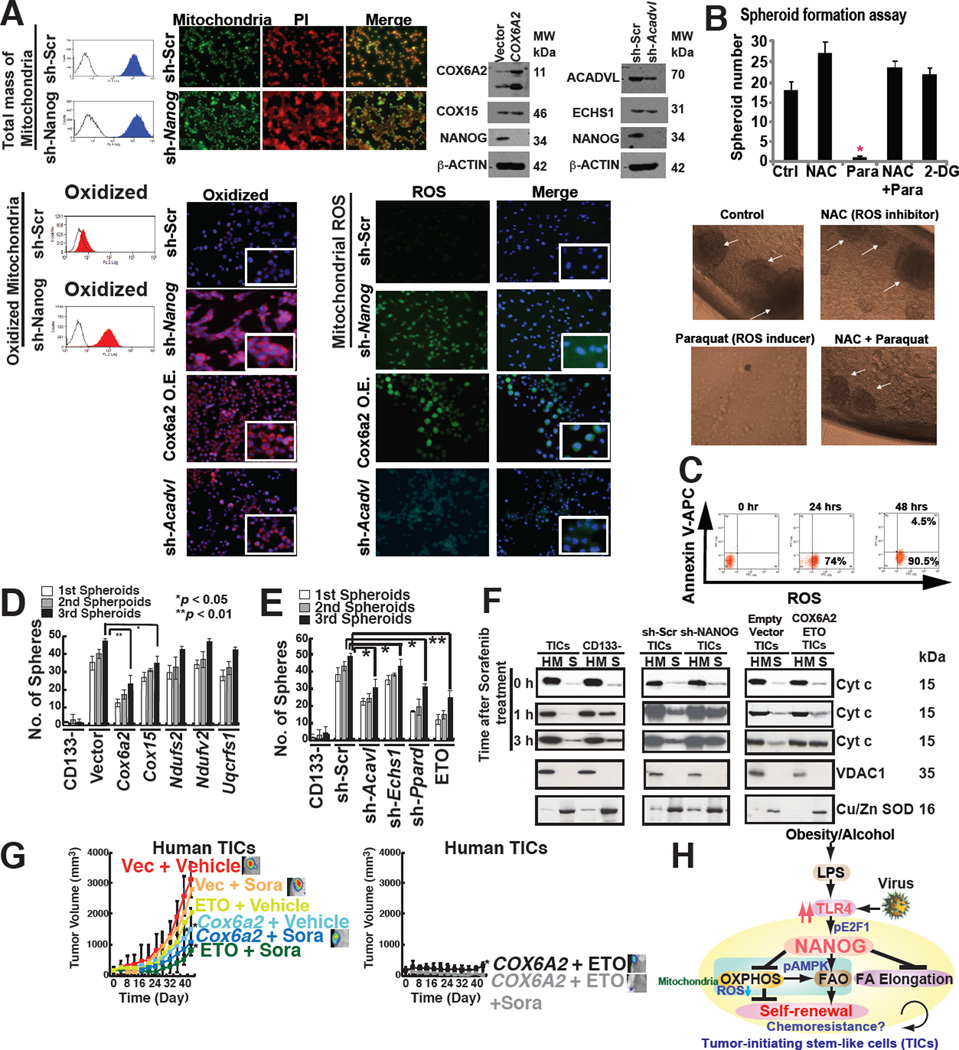
Stem cell markers, including NANOG, have been implicated in various cancers; however, the functional contribution of NANOG to cancer pathogenesis has remained unclear. Here, we show that NANOG is induced by Toll-like receptor 4 (TLR4) signaling via phosphorylation of E2F1 and that downregulation of Nanog slows down hepatocellular carcinoma (HCC) progression induced by alcohol western diet and hepatitis C virus protein in mice. NANOG ChIP-seq analyses reveal that NANOG regulates the expression of genes involved in mitochondrial metabolic pathways required to maintain tumor-initiating stem-like cells (TICs). NANOG represses mitochondrial oxidative phosphorylation (OXPHOS) genes, as well as ROS generation, and activates fatty acid oxidation (FAO) to support TIC self-renewal and drug resistance. Restoration of OXPHOS activity and inhibition of FAO renders TICs susceptible to a standard care chemotherapy drug for HCC, sorafenib. This study provides insights into the mechanisms of NANOG-mediated generation of TICs, tumorigenesis, and chemoresistance through reprogramming of mitochondrial metabolism.
Keywords: HCC; NANOG; OXPHOS; fatty acid; liver; metabolic reprogramming; self-renewal; tumor-initiating stem-like cells (TICs).
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
References
-
- Ferrick DA, Neilson A, Beeson C. Advances in measuring cellular bioenergetics using extracellular flux. Drug Discov Today. 2008;13:268–274. - PubMed
-
- Gutgesell A, Wen G, Konig B, Koch A, Spielmann J, Stangl GI, Eder K, Ringseis R. Mouse carnitine-acylcarnitine translocase (CACT) is transcriptionally regulated by PPARalpha and PPARdelta in liver cells. Biochim Biophys Acta. 2009;1790:1206–1216. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
