

Mechanism of FGF receptor dimerization and activation
- PMID: 26725515
- PMCID: PMC4725768
- DOI: 10.1038/ncomms10262
Mechanism of FGF receptor dimerization and activation
Abstract
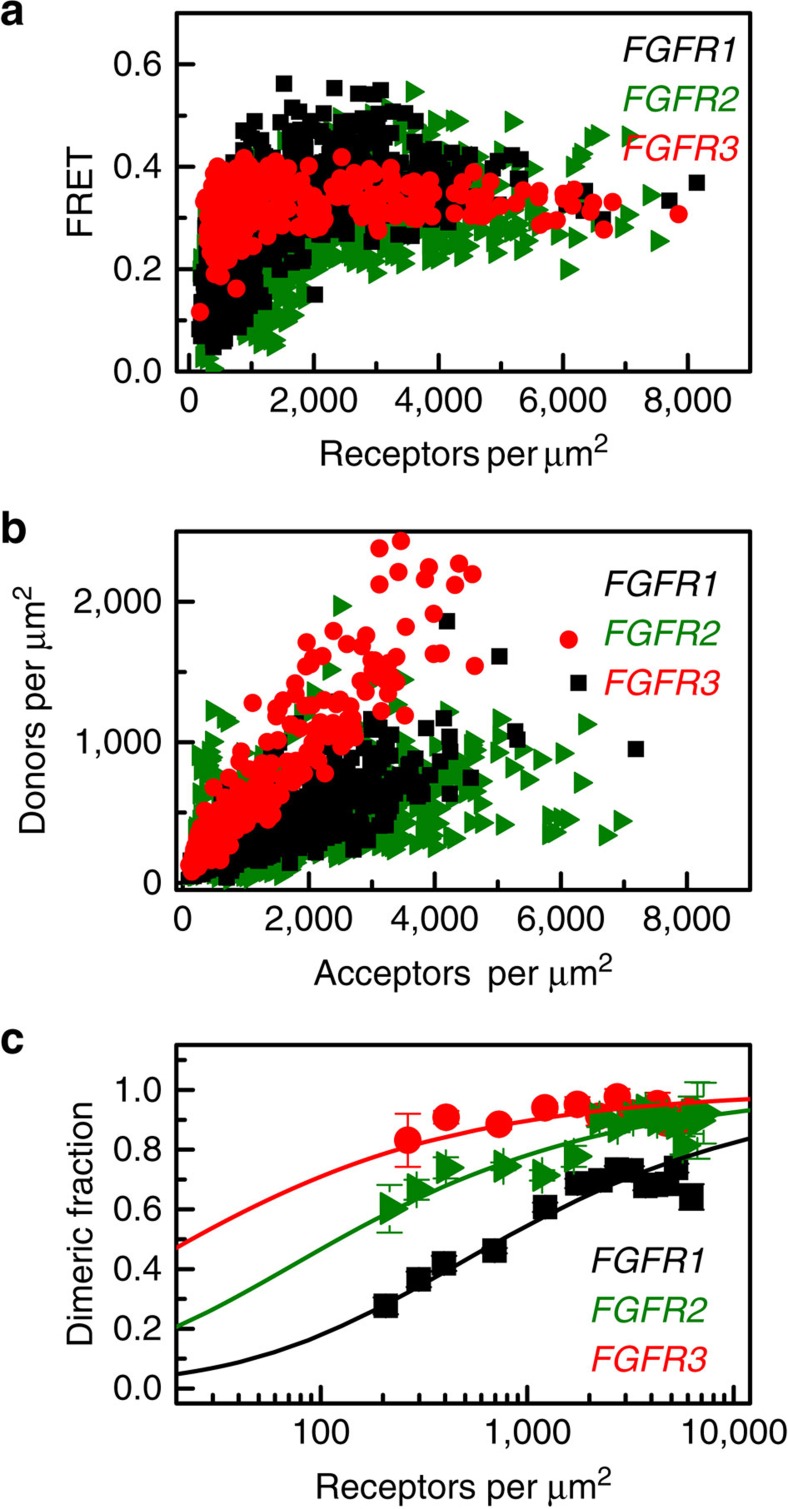
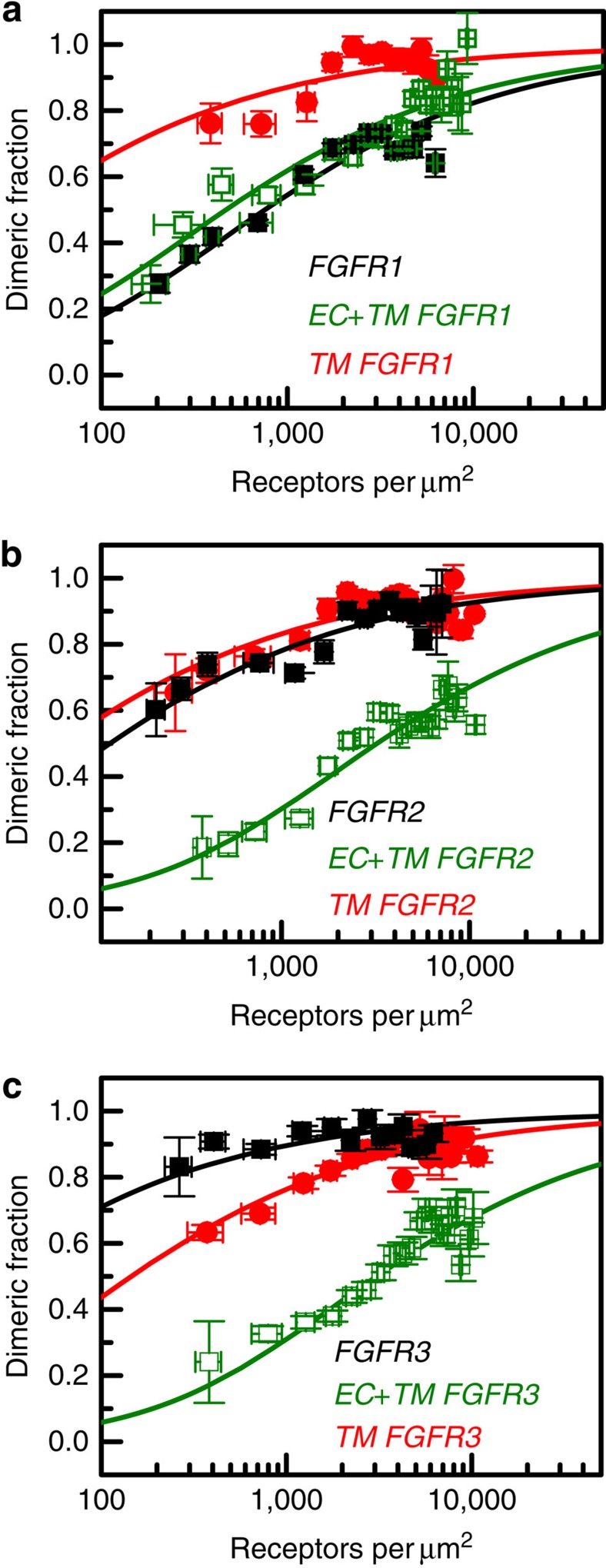
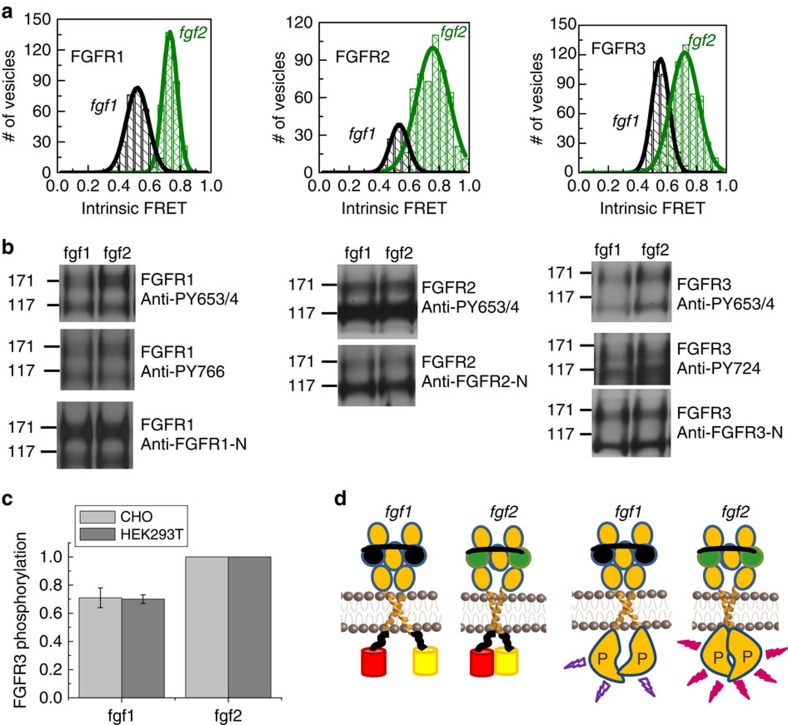
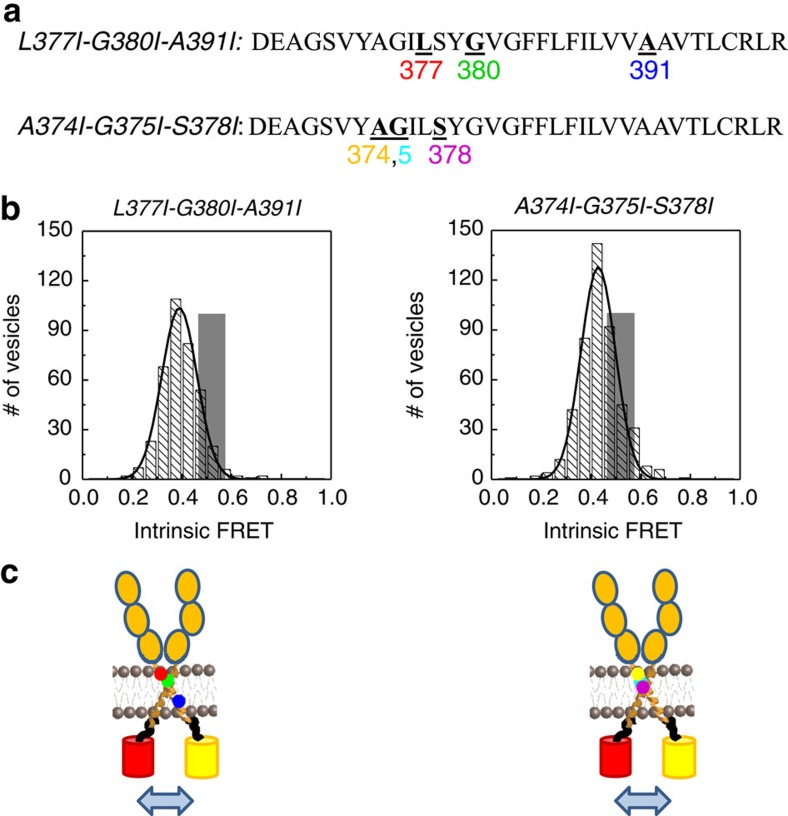
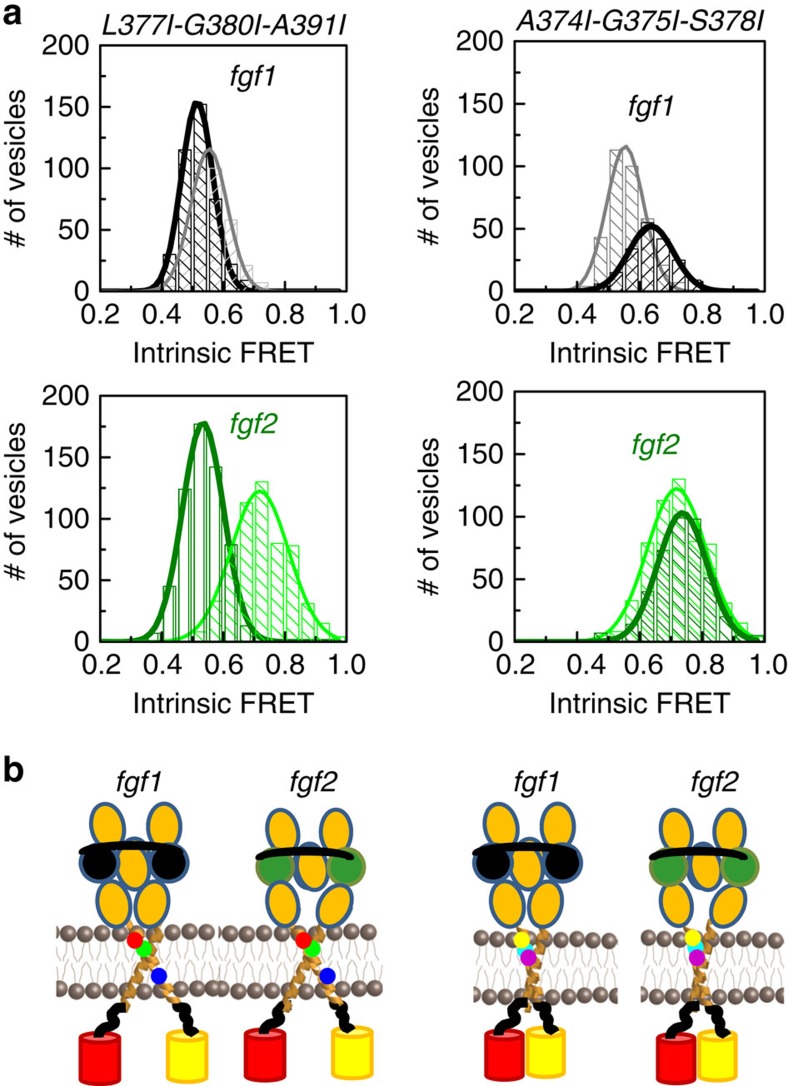
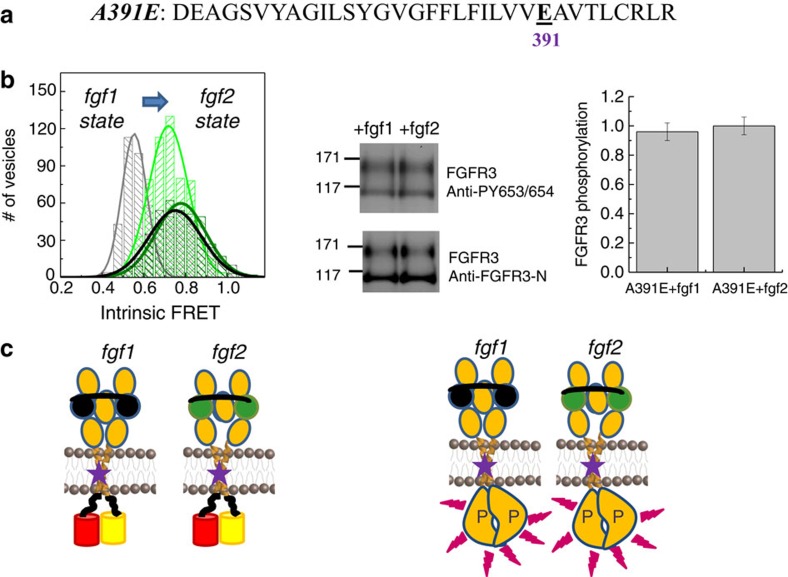
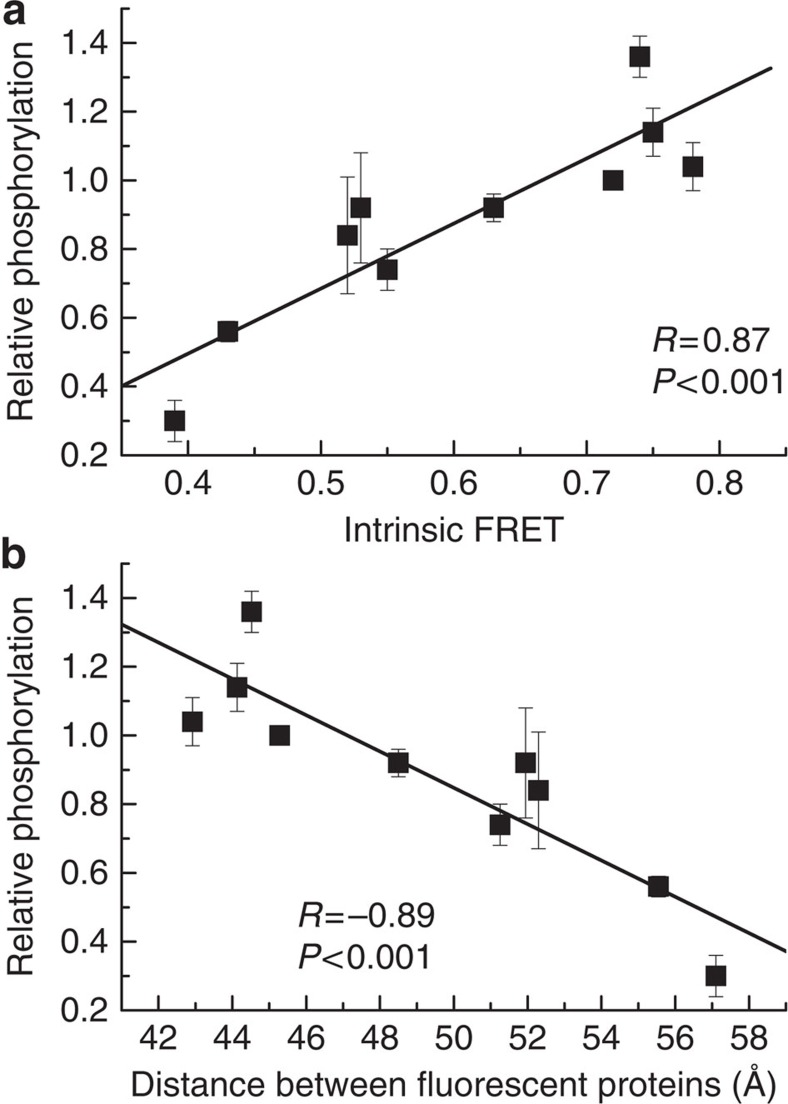
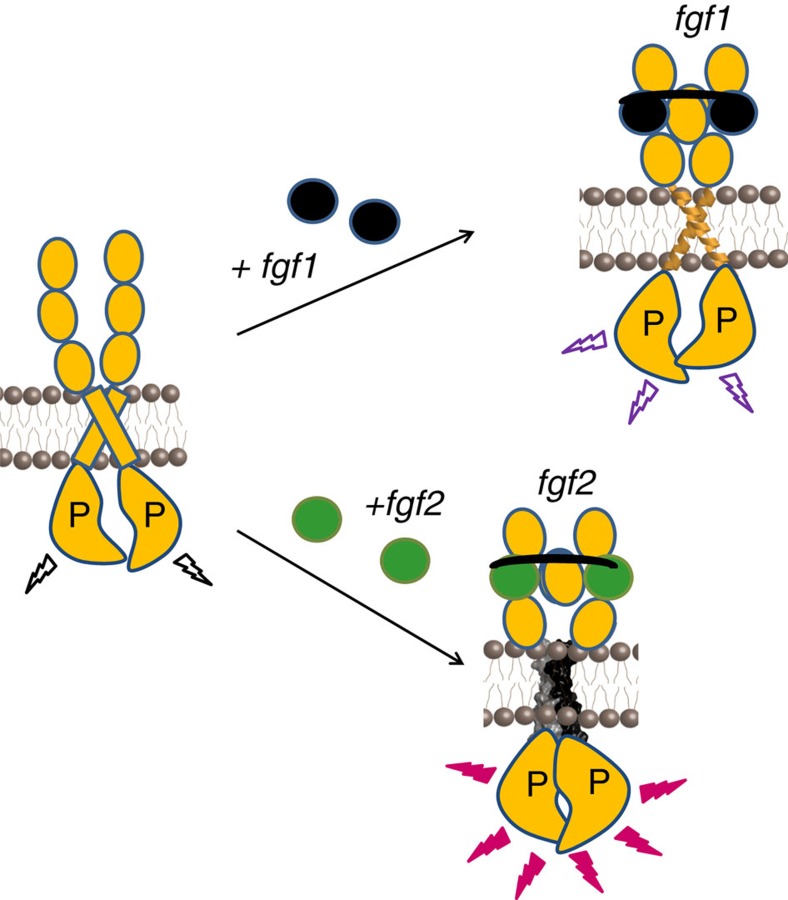
Fibroblast growth factors (fgfs) are widely believed to activate their receptors by mediating receptor dimerization. Here we show, however, that the FGF receptors form dimers in the absence of ligand, and that these unliganded dimers are phosphorylated. We further show that ligand binding triggers structural changes in the FGFR dimers, which increase FGFR phosphorylation. The observed effects due to the ligands fgf1 and fgf2 are very different. The fgf2-bound dimer structure ensures the smallest separation between the transmembrane (TM) domains and the highest possible phosphorylation, a conclusion that is supported by a strong correlation between TM helix separation in the dimer and kinase phosphorylation. The pathogenic A391E mutation in FGFR3 TM domain emulates the action of fgf2, trapping the FGFR3 dimer in its most active state. This study establishes the existence of multiple active ligand-bound states, and uncovers a novel molecular mechanism through which FGFR-linked pathologies can arise.
Figures
References
-
- Wilkie A. O. M., Morriss-Kay G. M., Jones E. Y. & Heath J. K. Functions of fibroblast growth factors and their receptors. Curr. Biol. 5, 500–507 (1995). - PubMed
-
- Eswarakumar V. P., Lax I. & Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 16, 139–149 (2005). - PubMed
-
- Ornitz D. M. FGFs, heparan sulfate and FGFRs: complex interactions essential for development. BioEssays 22, 108–112 (2000). - PubMed
-
- Deng C., Wynshaw-Boris A., Zhou F., Kuo A. & Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell 84, 911–921 (1996). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
