

doi: 10.1016/j.trecan.2015.10.007.
Deciphering the Code of the Cancer Genome: Mechanisms of Chromosome Rearrangement
Affiliations
- PMID: 26726318
- PMCID: PMC4695301
- DOI: 10.1016/j.trecan.2015.10.007
Item in Clipboard
Deciphering the Code of the Cancer Genome: Mechanisms of Chromosome Rearrangement
Trends Cancer.
.
Abstract
Chromosome rearrangement plays a causal role in tumorigenesis by contributing to the inactivation of tumor suppressor genes, the dysregulated expression or amplification of oncogenes and the generation of novel gene fusions. Chromosome breaks are important intermediates in this process. How, when and where these breaks arise and the specific mechanisms engaged in their repair strongly influence the resulting patterns of chromosome rearrangement. Here, we review recent progress in understanding how certain distinctive features of the cancer genome, including clustered mutagenesis, tandem segmental duplications, complex breakpoints, chromothripsis, chromoplexy and chromoanasynthesis may arise.
Figures
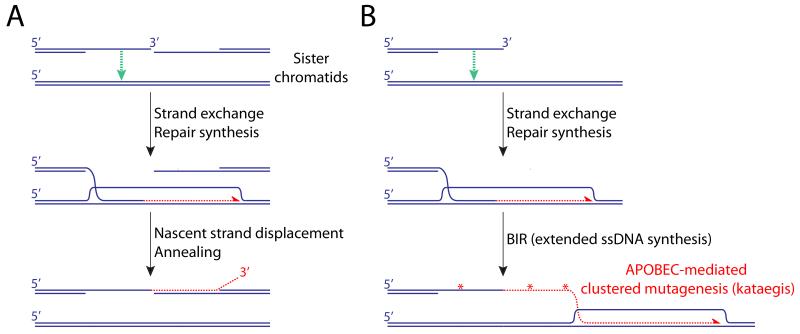
HR mediators (primarily BRCA2 in mammalian cells and Rad52 in yeast) displace RPA from ssDNA on the resected DNA end and load Rad51, forming a nucleoprotein filament. The Rad51 filament performs a homology-seeking invasion of neighboring double stranded (ds)DNA molecules and, if a high degree of homology is detected (typically ≥100bp), a DNA polymerase extends the 3′ end of the invading (“nascent”) strand. Sequence differences copied from the donor DNA alter the sequence of the repaired DNA molecule (“gene conversion”). In S/G2 phase, the neighboring sister chromatid is the preferred donor for recombination, with potential for error-free DSB repair. However, the Rad51 filament can also detect homology at distant loci, potentially contributing to genome rearrangement. A. “Synthesis-dependent strand annealing” (SDSA) pathway of HR. Rad51-mediated strand exchange (green arrow) enables repair synthesis (red half-arrow), using the donor as template. In somatic cells, HR termination entails helicase-driven displacement of the nascent strand from the donor template followed by annealing (homologous pairing) with complementary ssDNA of the second end of the DSB. This termination mechanism does not lead to crossing over. B. Break-induced replication and kataegis. Typically triggered by a one-ended invasion, BIR is mediated by a “migrating bubble” mechanisms of leading strand synthesis (red half-arrow). The extensive tracts of ssDNA generated by BIR are potential targets of cytidine deamination (red asterisks) by APOBEC family enzymes, leading to patterns of clustered mutagenesis (kataegis).
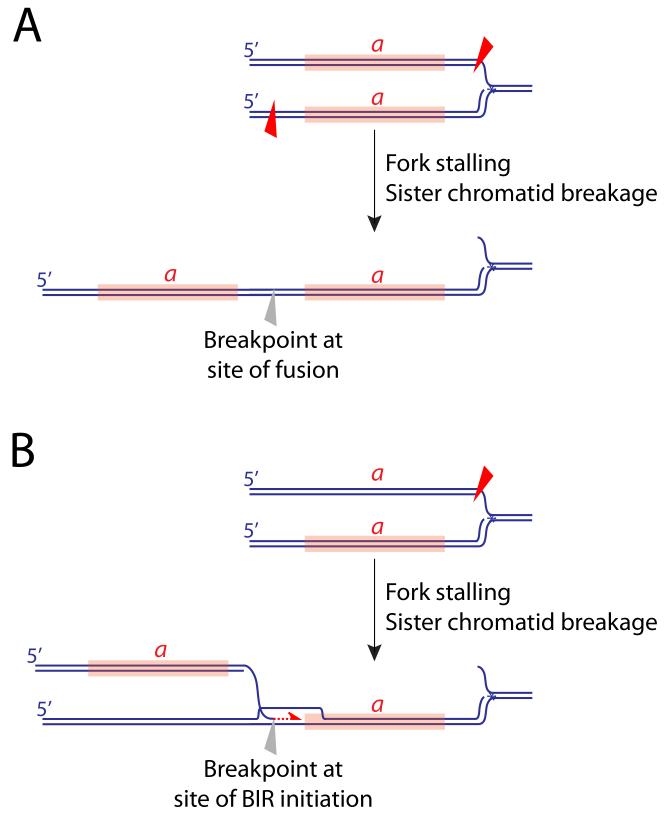
Aberrant processing of a stalled fork, perhaps augmented by an HR defect, may produce unscheduled breakage of sister chromatids in the vicinity of the stalled fork. Tandem duplication of segment a (marked in orange) could arise by asymmetrical breakage (red triangles) and fusion of sister chromatids (A), or by break-induced replication following breakage of one sister chromatid (B).
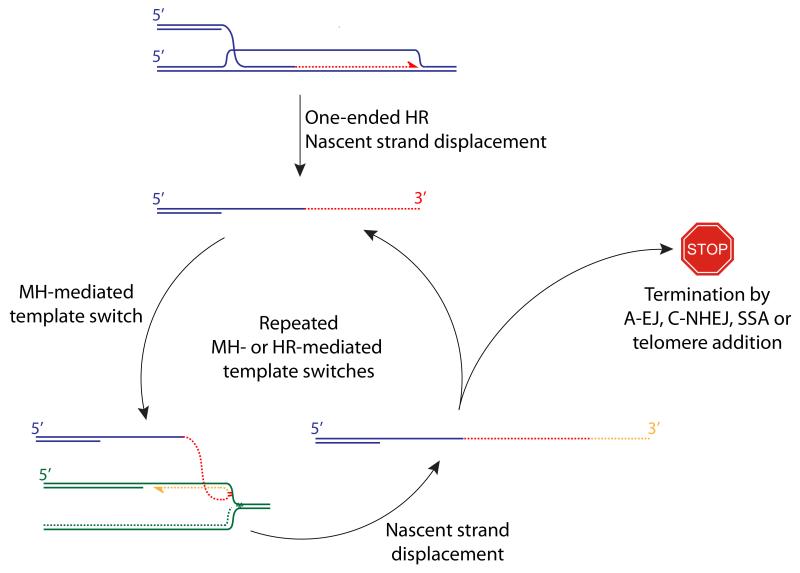
The displaced 3′ ssDNA tail produced following a “one-ended” HR invasion may undergo MH-mediated template switching—shown here into the lagging strand of a neighboring stalled fork. Repeated rounds of MH- or HR-mediated template switches could generate the complex breakpoints observed in cancer. Termination of a template switch cycle must involve either joining to a second DNA end or telomere addition.
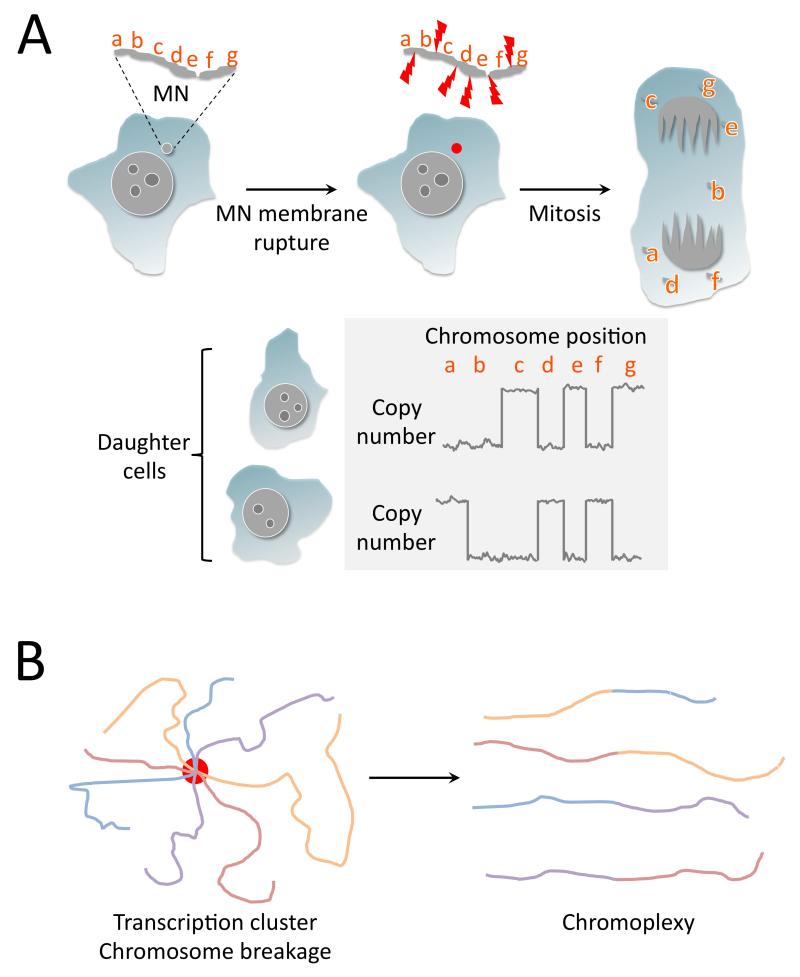
A. Chromothripsis can arise following mitotic damage to micronucleated chromosome(s). A micronucleated chromosome (“MN”) containing loci “a-g” is exposed to cytoplasmic nucleases when the MN membrane ruptures (red dot) and undergoes fragmentation. During the subsequent mitosis, the chromothriptic fragments segregate randomly into the two daughter cells, or are lost (e.g., fragment b shown here). The daughter cell genomes reveal constrained copy number oscillations characteristic of chromothripsis. B. A model of chromoplexy in prostate cancer. Clustering of transcriptional elements (red circle), for example, at androgen receptor (AR) responsive loci, coupled with AR-associated chromosome breakage, leads to rejoining of broken chromosoms with the production of linked translocations characteristic of chromoplexy.
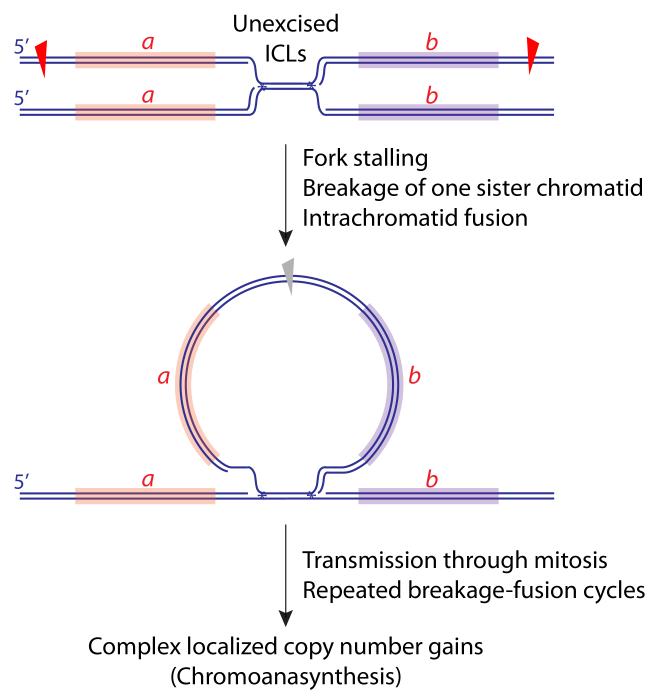
An unexcised ICL could lead to breakage of one sister chromatid, with circularization of a retained fragment, containing loci a and b, as shown. Provided the retained circular fragment lacks a centromere, it will not be a barrier to mitosis and will carry additional copies of loci a and b into the subsequent S phase. Scheduled replication, rearrangement and integration of the fragment into the gemome could contribute to the localized copy number gains characteristic of chromoanasynthesis. Notably, if the circularized fragment were to contain an origin of replication (not shown), this could further increase the opportunity for rapid copy number gains via rolling circle replication (not shown).
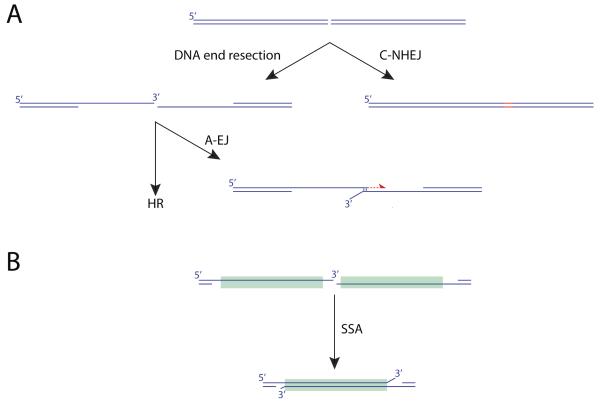
(associated with Box1): Diagram of major pathways involved in the repair of DNA double strand breaks (HR, C-NHEJ, A-EJ, SSA).
References
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. - PubMed
-
- Cahill DP, Kinzler KW, Vogelstein B, Lengauer C. Genetic instability and darwinian selection in tumours. Trends in cell biology. 1999;9:M57–60. - PubMed
-
- Neelsen KJ, Lopes M. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol. 2015;16:207–220. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources