

The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux
- PMID: 26727229
- PMCID: PMC4701542
- DOI: 10.1172/JCI77812
The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux
Abstract
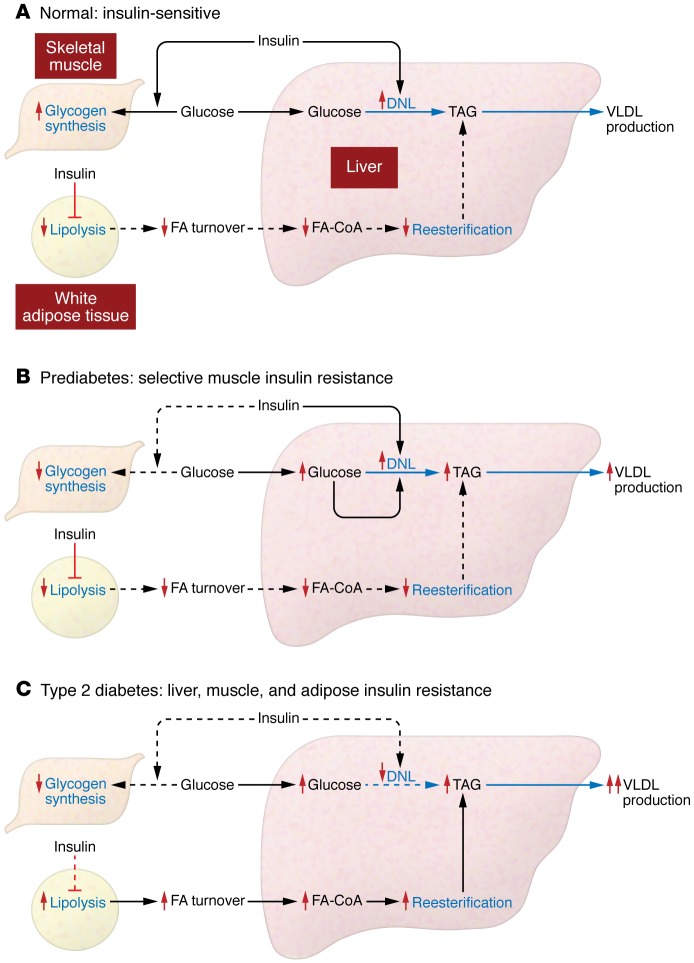
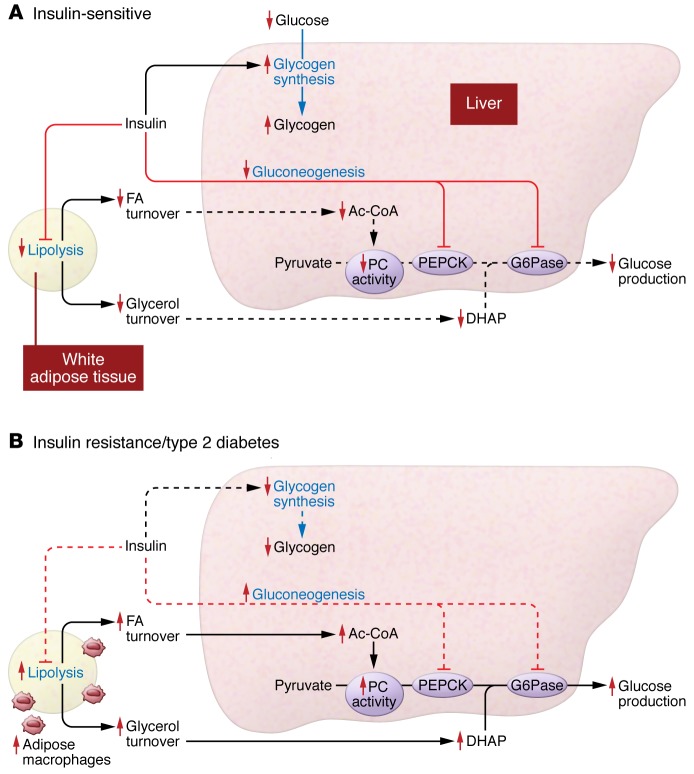
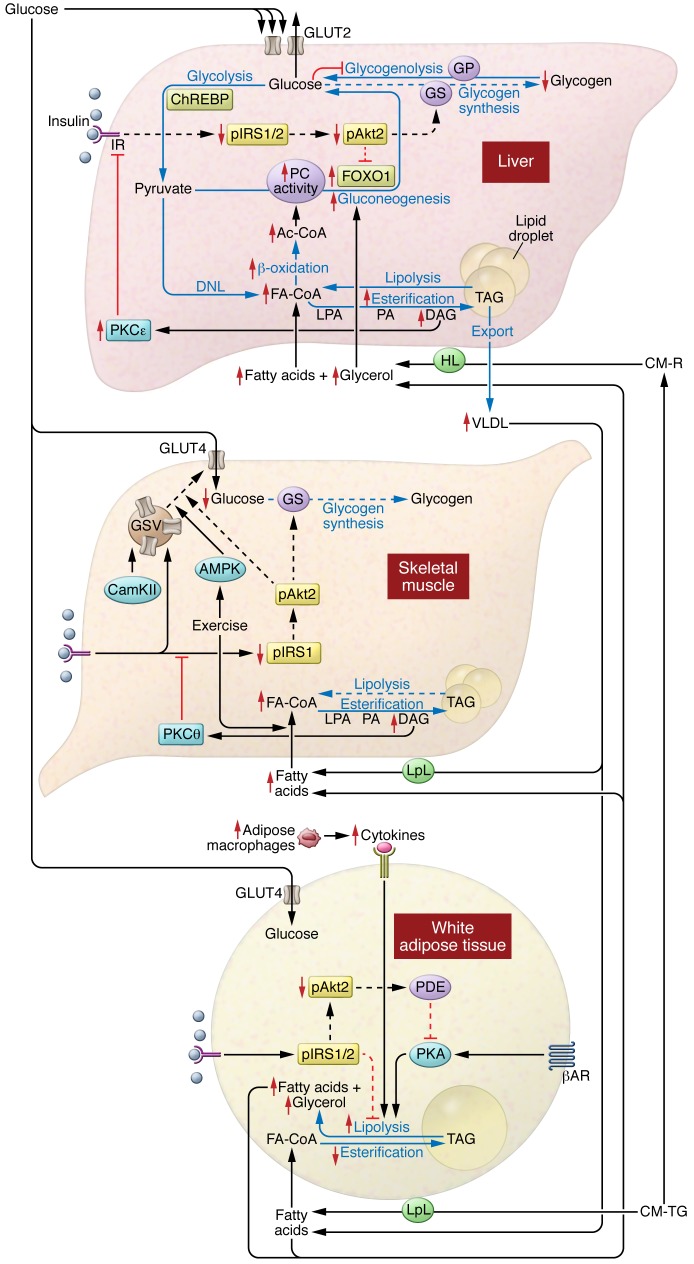
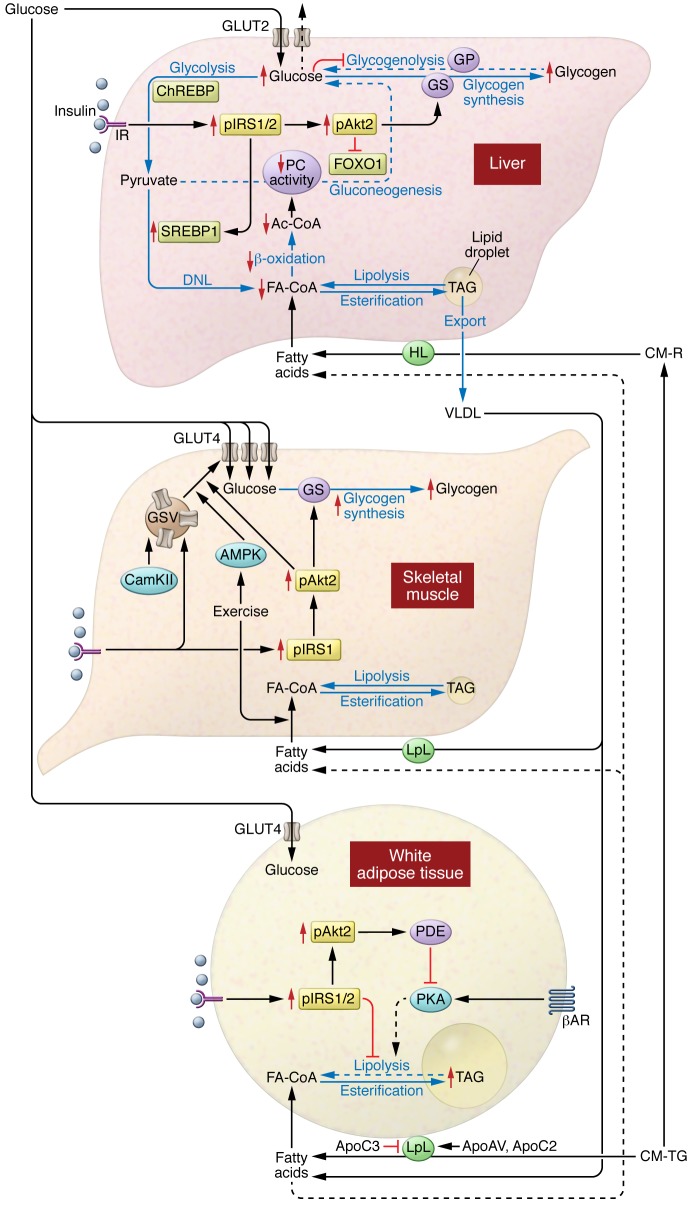
Insulin resistance arises when the nutrient storage pathways evolved to maximize efficient energy utilization are exposed to chronic energy surplus. Ectopic lipid accumulation in liver and skeletal muscle triggers pathways that impair insulin signaling, leading to reduced muscle glucose uptake and decreased hepatic glycogen synthesis. Muscle insulin resistance, due to ectopic lipid, precedes liver insulin resistance and diverts ingested glucose to the liver, resulting in increased hepatic de novo lipogenesis and hyperlipidemia. Subsequent macrophage infiltration into white adipose tissue (WAT) leads to increased lipolysis, which further increases hepatic triglyceride synthesis and hyperlipidemia due to increased fatty acid esterification. Macrophage-induced WAT lipolysis also stimulates hepatic gluconeogenesis, promoting fasting and postprandial hyperglycemia through increased fatty acid delivery to the liver, which results in increased hepatic acetyl-CoA content, a potent activator of pyruvate carboxylase, and increased glycerol conversion to glucose. These substrate-regulated processes are mostly independent of insulin signaling in the liver but are dependent on insulin signaling in WAT, which becomes defective with inflammation. Therapies that decrease ectopic lipid storage and diminish macrophage-induced WAT lipolysis will reverse the root causes of type 2 diabetes.
Figures
References
-
- Kasvinsky PJ, Shechosky S, Fletterick RJ. Synergistic regulation of phosphorylase a by glucose and caffeine. J Biol Chem. 1978;253(24):9102–9106. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
