

Neuroprotection by selective neuronal deletion of Atg7 in neonatal brain injury
- PMID: 26727396
- PMCID: PMC4835980
- DOI: 10.1080/15548627.2015.1132134
Neuroprotection by selective neuronal deletion of Atg7 in neonatal brain injury
Abstract
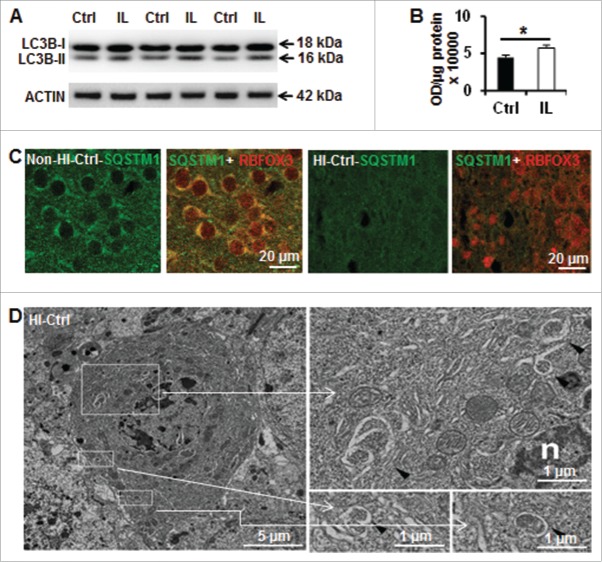
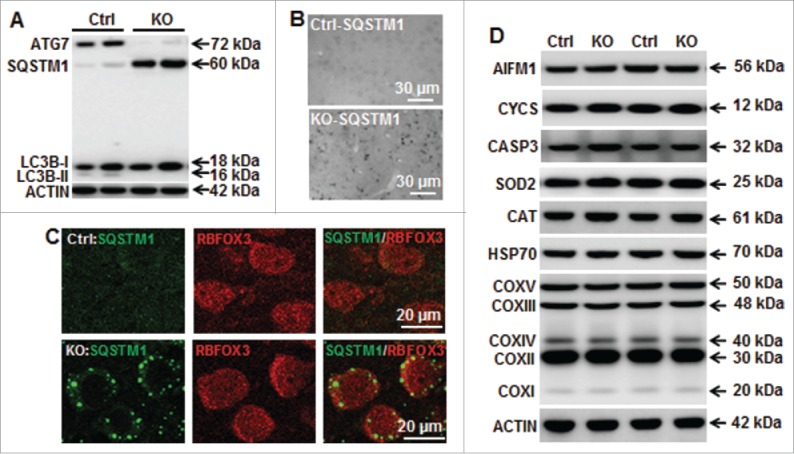
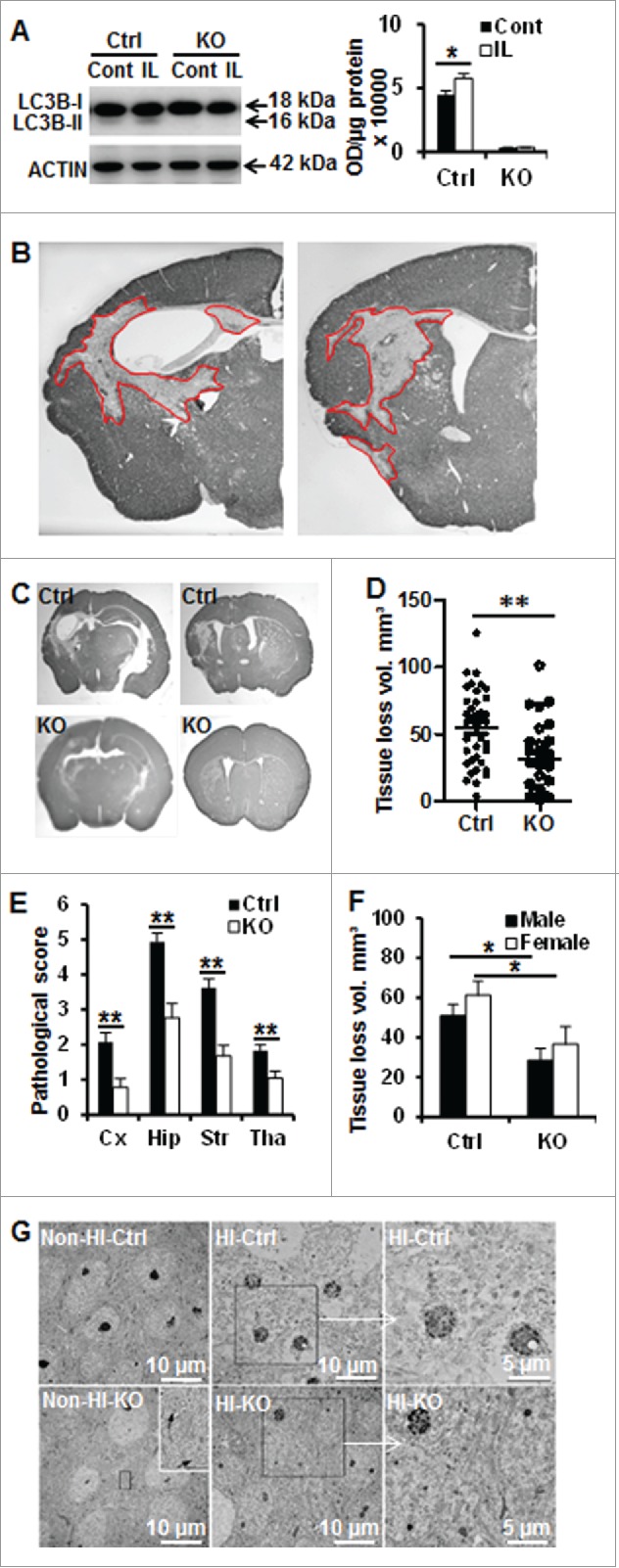
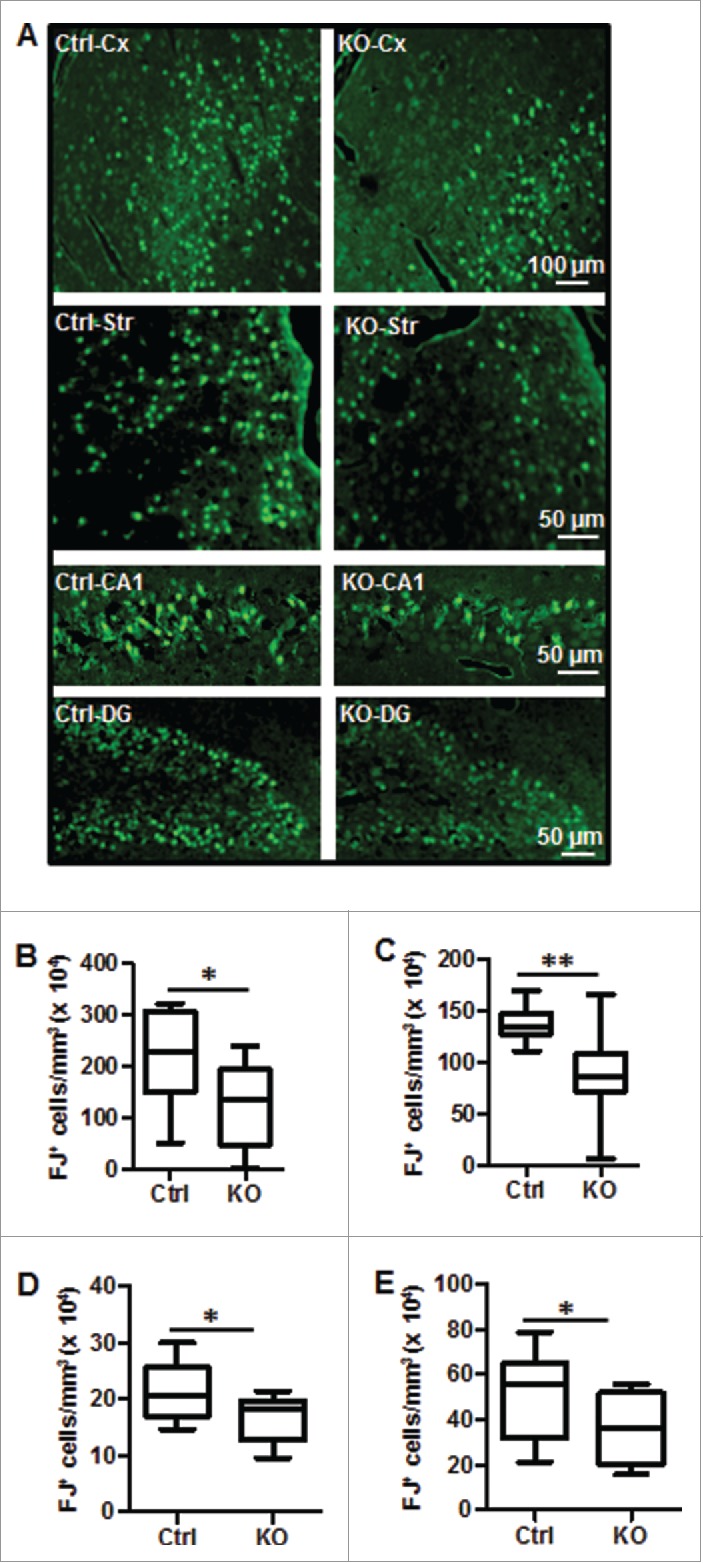
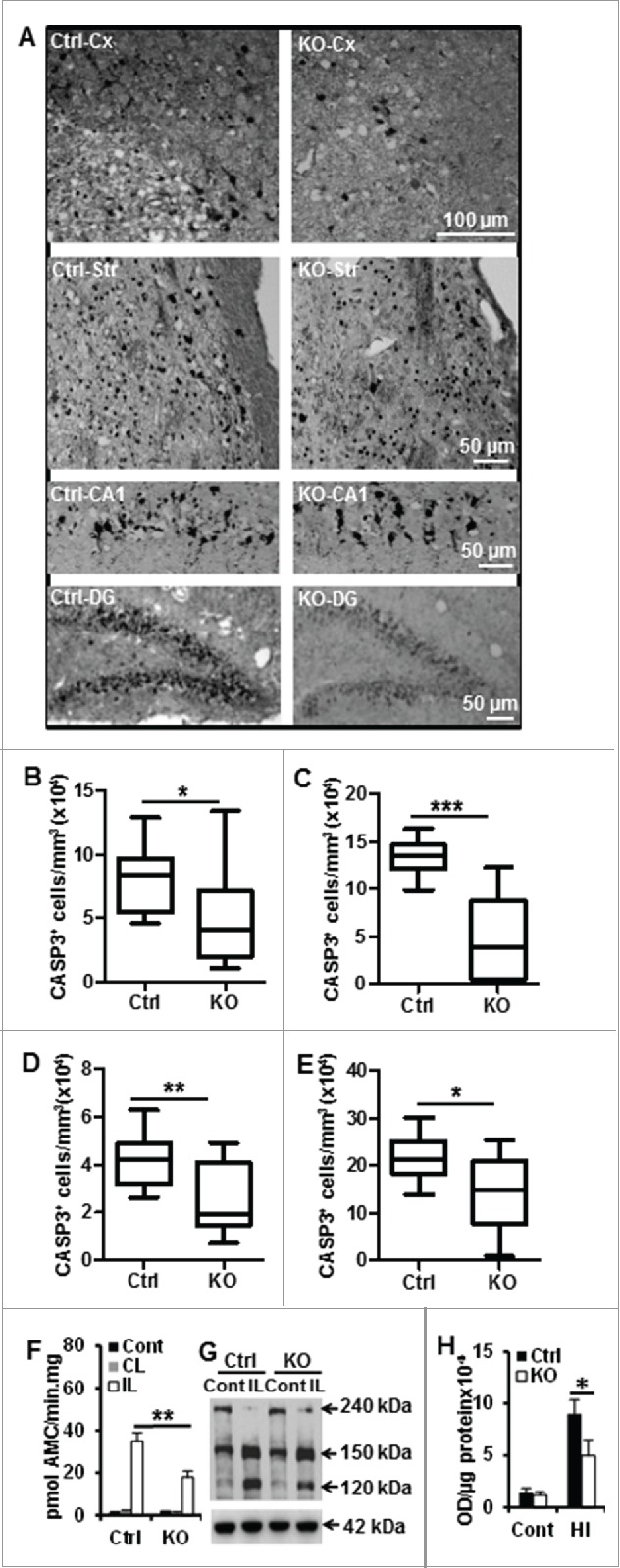
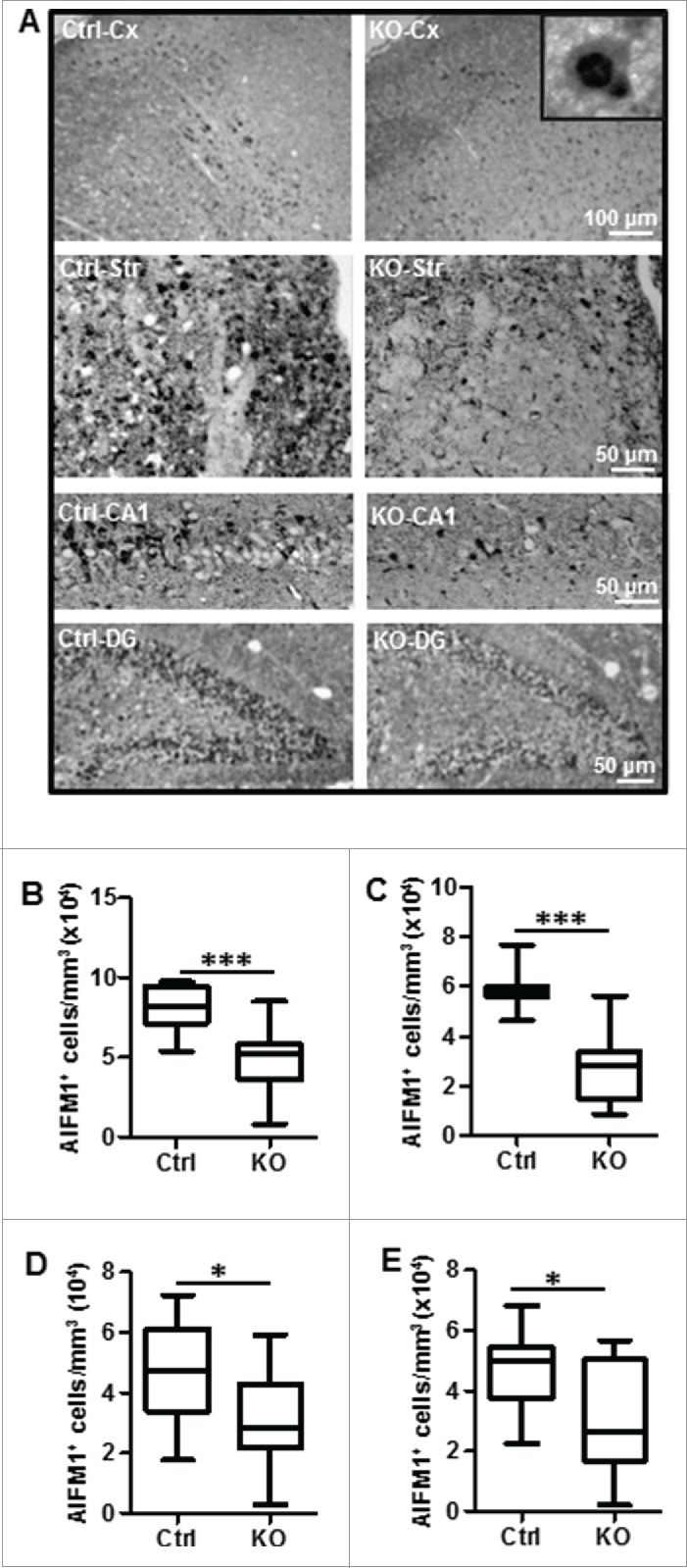
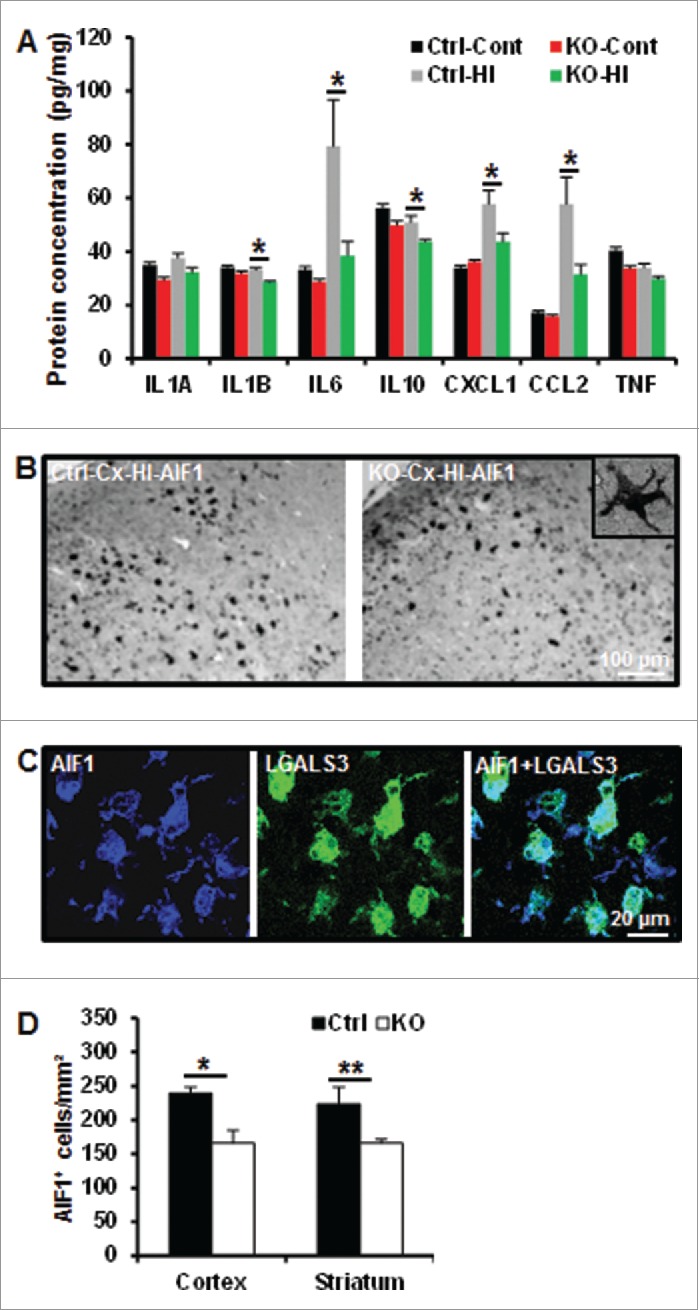
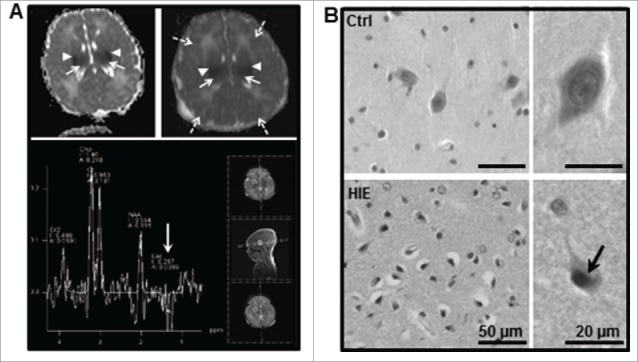
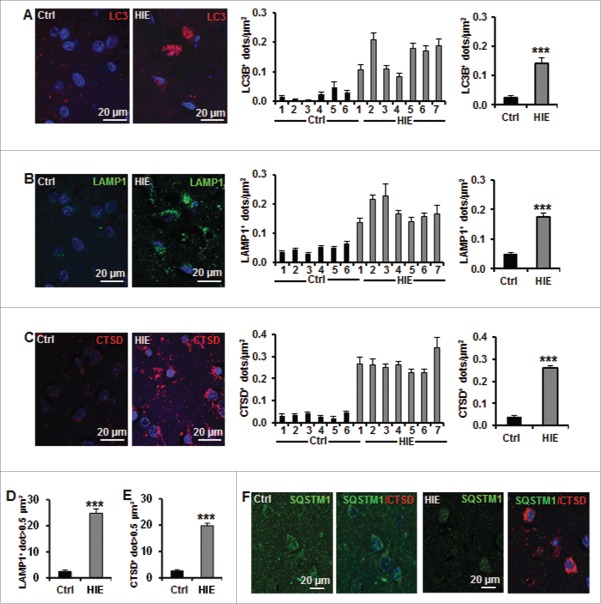
Perinatal asphyxia induces neuronal cell death and brain injury, and is often associated with irreversible neurological deficits in children. There is an urgent need to elucidate the neuronal death mechanisms occurring after neonatal hypoxia-ischemia (HI). We here investigated the selective neuronal deletion of the Atg7 (autophagy related 7) gene on neuronal cell death and brain injury in a mouse model of severe neonatal hypoxia-ischemia. Neuronal deletion of Atg7 prevented HI-induced autophagy, resulted in 42% decrease of tissue loss compared to wild-type mice after the insult, and reduced cell death in multiple brain regions, including apoptosis, as shown by decreased caspase-dependent and -independent cell death. Moreover, we investigated the lentiform nucleus of human newborns who died after severe perinatal asphyxia and found increased neuronal autophagy after severe hypoxic-ischemic encephalopathy compared to control uninjured brains, as indicated by the numbers of MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3)-, LAMP1 (lysosomal-associated membrane protein 1)-, and CTSD (cathepsin D)-positive cells. These findings reveal that selective neuronal deletion of Atg7 is strongly protective against neuronal death and overall brain injury occurring after HI and suggest that inhibition of HI-enhanced autophagy should be considered as a potential therapeutic target for the treatment of human newborns developing severe hypoxic-ischemic encephalopathy.
Keywords: ATG7; apoptosis; autophagy; caspase; hypoxic-ischemic encephalopathy; newborn.
Figures
References
-
- Mwaniki MK, Atieno M, Lawn JE, Newton CR. Long-term neurodevelopmental outcomes after intrauterine and neonatal insults: a systematic review. Lancet 2012; 379:445-52; PMID:22244654; http://dx.doi.org/ 10.1016/S0140-6736(11)61577-8 - DOI - PMC - PubMed
-
- Krageloh-Mann I, Helber A, Mader I, Staudt M, Wolff M, Groenendaal F, DeVries L. Bilateral lesions of thalamus and basal ganglia: origin and outcome. Dev Med Child Neurol 2002; 44:477-84; PMID:12162385; http://dx.doi.org/ 10.1111/j.1469-8749.2002.tb00309.x - DOI - PubMed
-
- Northington FJ, Chavez-Valdez R, Martin LJ. Neuronal cell death in neonatal hypoxia-ischemia. Ann Neurol 2011; 69:743-58; PMID:21520238; http://dx.doi.org/ 10.1002/ana.22419 - DOI - PMC - PubMed
-
- Carlsson Y, Schwendimann L, Vontell R, Rousset CI, Wang X, Lebon S, Charriaut-Marlangue C, Supramaniam V, Hagberg H, Gressens P, et al.. Genetic inhibition of caspase-2 reduces hypoxic-ischemic and excitotoxic neonatal brain injury. Ann Neurol 2011; 70:781-9; PMID:21674587; http://dx.doi.org/ 10.1002/ana.22431 - DOI - PubMed
-
- Zhu C, Wang X, Deinum J, Huang Z, Gao J, Modjtahedi N, Neagu MR, Nilsson M, Eriksson PS, Hagberg H, et al.. Cyclophilin A participates in the nuclear translocation of apoptosis-inducing factor in neurons after cerebral hypoxia-ischemia. J Exp Med 2007; 204:1741-8; PMID:17635954; http://dx.doi.org/ 10.1084/jem.20070193 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous