

Quantifying influenza virus diversity and transmission in humans
- PMID: 26727660
- PMCID: PMC4731279
- DOI: 10.1038/ng.3479
Quantifying influenza virus diversity and transmission in humans
Abstract
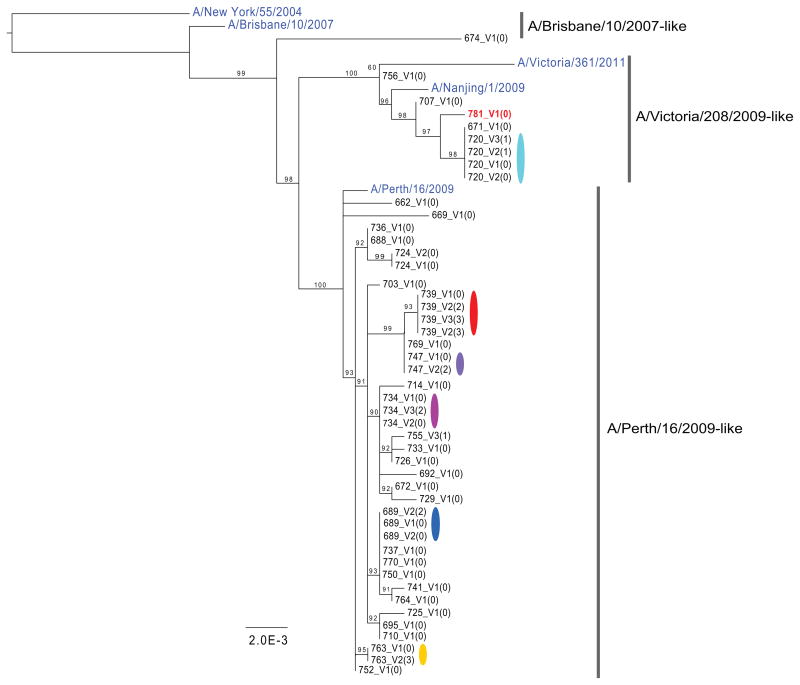
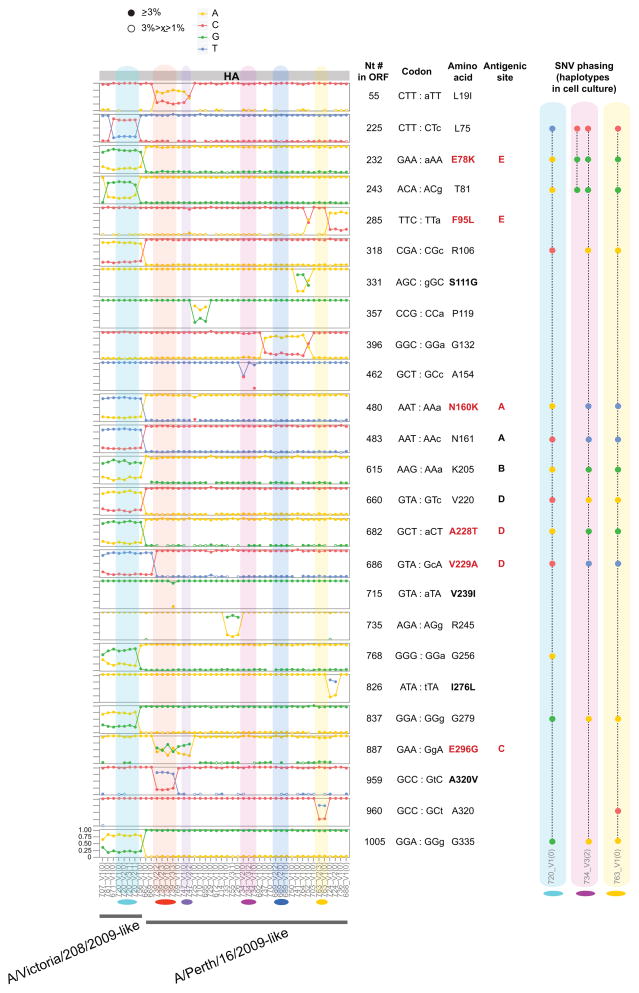
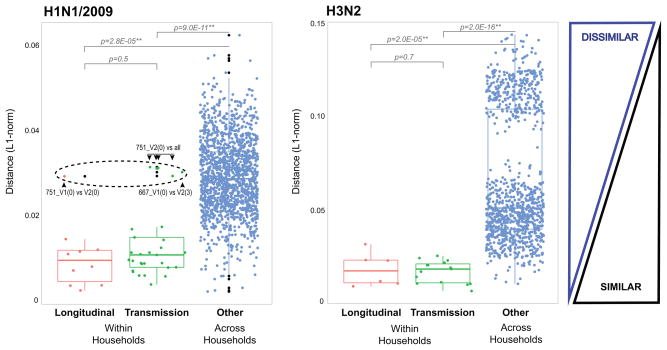
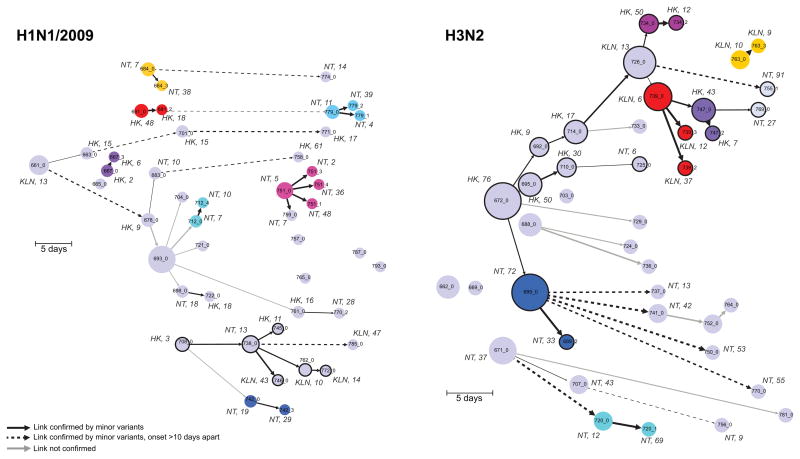
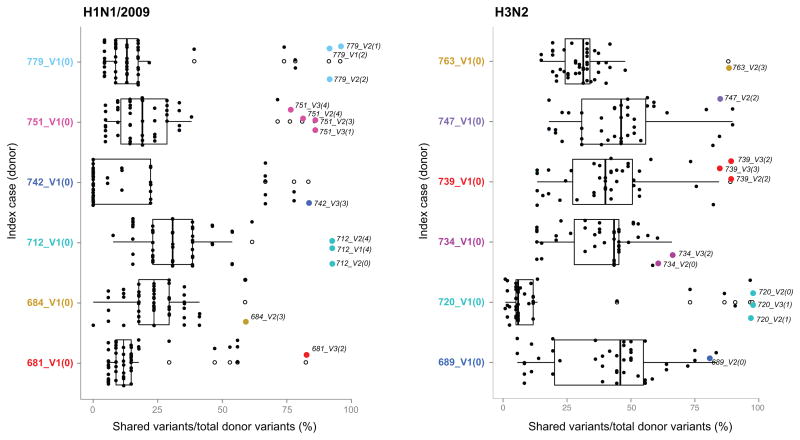
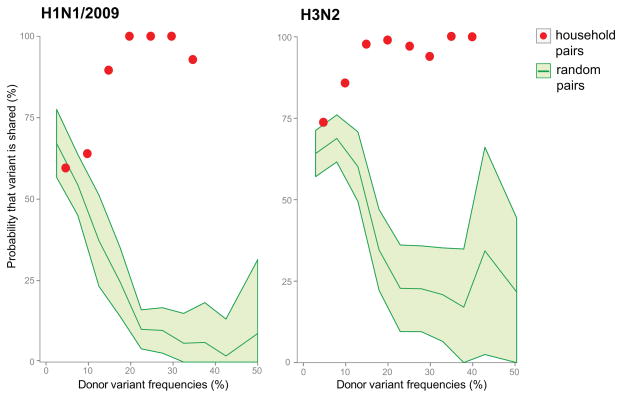
Influenza A virus is characterized by high genetic diversity. However, most of what is known about influenza evolution has come from consensus sequences sampled at the epidemiological scale that only represent the dominant virus lineage within each infected host. Less is known about the extent of within-host virus diversity and what proportion of this diversity is transmitted between individuals. To characterize virus variants that achieve sustainable transmission in new hosts, we examined within-host virus genetic diversity in household donor-recipient pairs from the first wave of the 2009 H1N1 pandemic when seasonal H3N2 was co-circulating. Although the same variants were found in multiple members of the community, the relative frequencies of variants fluctuated, with patterns of genetic variation more similar within than between households. We estimated the effective population size of influenza A virus across donor-recipient pairs to be approximately 100-200 contributing members, which enabled the transmission of multiple lineages, including antigenic variants.
Conflict of interest statement
Competing financial interests:
Authors have no competing financial interests.
Figures
Comment in
-
Reconciling disparate estimates of viral genetic diversity during human influenza infections.Nat Genet. 2019 Sep;51(9):1298-1301. doi: 10.1038/s41588-019-0349-3. Nat Genet. 2019. PMID: 30804564 Free PMC article. No abstract available.
-
Reply to 'Reconciling disparate estimates of viral genetic diversity during human influenza infections'.Nat Genet. 2019 Sep;51(9):1301-1303. doi: 10.1038/s41588-019-0486-8. Nat Genet. 2019. PMID: 31406345 No abstract available.
References
-
- Bush RM, Fitch WM, Bender CA, Cox NJ. Positive selection on the H3 hemagglutinin gene of human influenza virus A. Mol Biol Evol. 1999;16:1457–65. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
