

The autism-associated MET receptor tyrosine kinase engages early neuronal growth mechanism and controls glutamatergic circuits development in the forebrain
- PMID: 26728565
- PMCID: PMC4914424
- DOI: 10.1038/mp.2015.182
The autism-associated MET receptor tyrosine kinase engages early neuronal growth mechanism and controls glutamatergic circuits development in the forebrain
Abstract
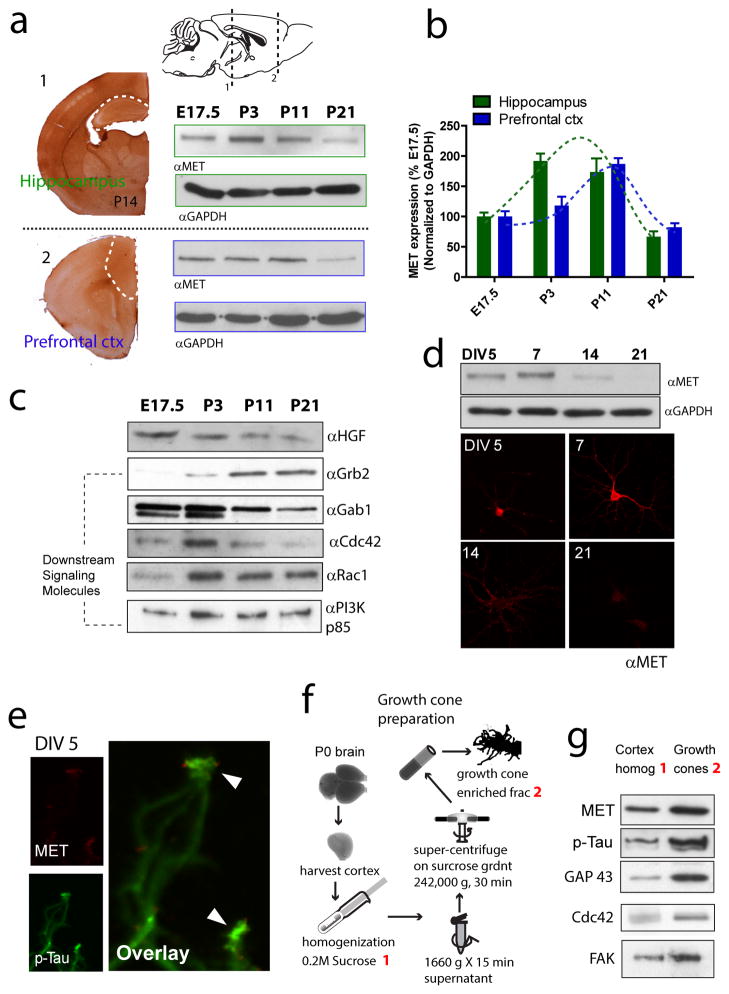
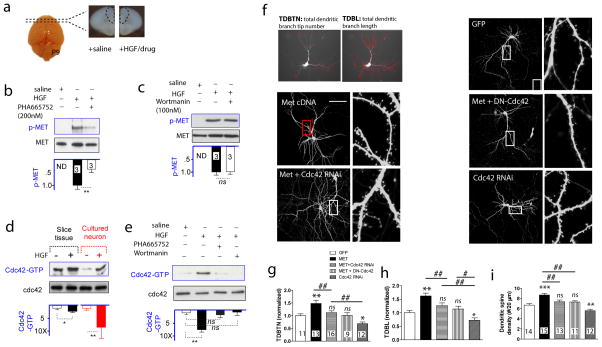
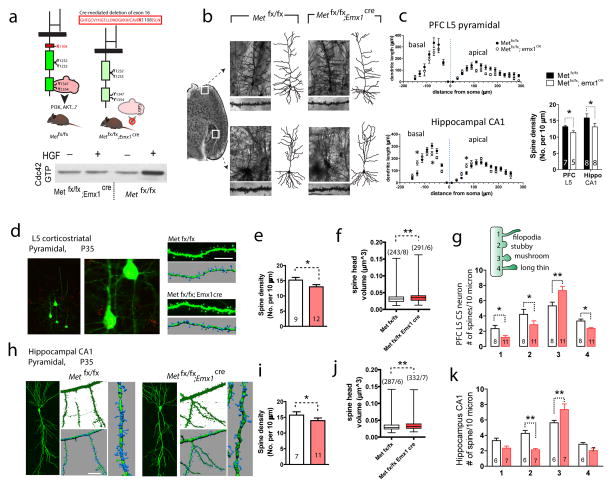
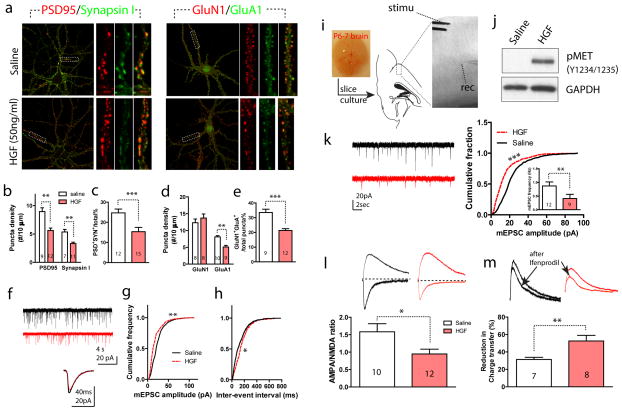
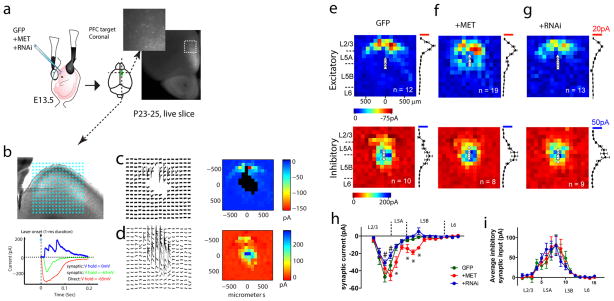
The human MET gene imparts a replicated risk for autism spectrum disorder (ASD), and is implicated in the structural and functional integrity of brain. MET encodes a receptor tyrosine kinase, MET, which has a pleiotropic role in embryogenesis and modifies a large number of neurodevelopmental events. Very little is known, however, on how MET signaling engages distinct cellular events to collectively affect brain development in ASD-relevant disease domains. Here, we show that MET protein expression is dynamically regulated and compartmentalized in developing neurons. MET is heavily expressed in neuronal growth cones at early developmental stages and its activation engages small GTPase Cdc42 to promote neuronal growth, dendritic arborization and spine formation. Genetic ablation of MET signaling in mouse dorsal pallium leads to altered neuronal morphology indicative of early functional maturation. In contrast, prolonged activation of MET represses the formation and functional maturation of glutamatergic synapses. Moreover, manipulating MET signaling levels in vivo in the developing prefrontal projection neurons disrupts the local circuit connectivity made onto these neurons. Therefore, normal time-delimited MET signaling is critical in regulating the timing of neuronal growth, glutamatergic synapse maturation and cortical circuit function. Dysregulated MET signaling may lead to pathological changes in forebrain maturation and connectivity, and thus contribute to the emergence of neurological symptoms associated with ASD.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013;14(1):7–23. - PubMed
-
- Uehara Y, Minowa O, Mori C, Shiota K, Kuno J, Noda T, et al. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature. 1995;373(6516):702–705. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
