

Evidence-Based Theory for Integrated Genome Regulation of Ontogeny--An Unprecedented Role of Nuclear FGFR1 Signaling
- PMID: 26729628
- PMCID: PMC5067692
- DOI: 10.1002/jcp.25298
Evidence-Based Theory for Integrated Genome Regulation of Ontogeny--An Unprecedented Role of Nuclear FGFR1 Signaling
Abstract
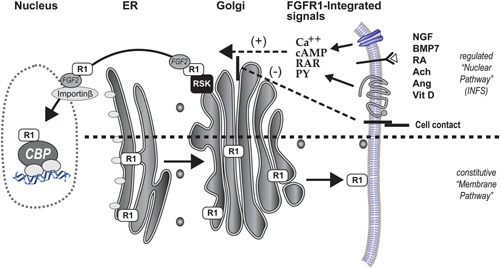
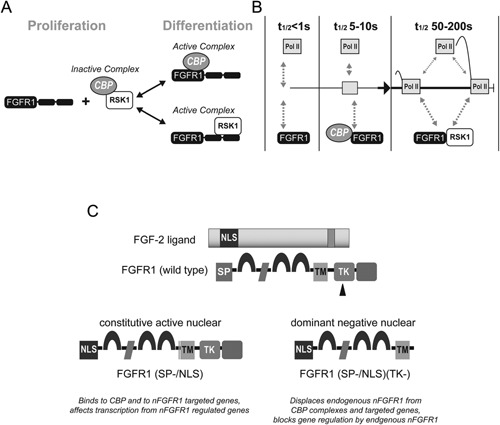
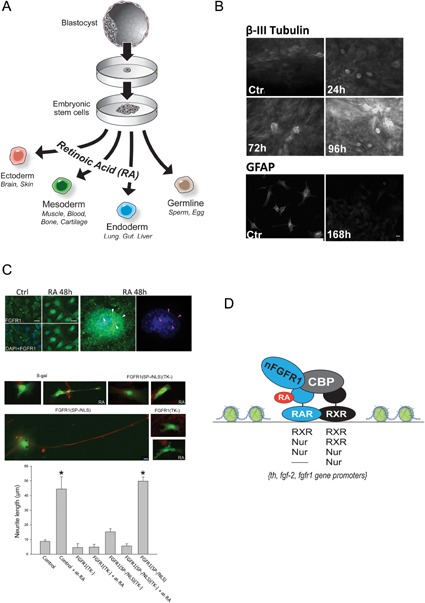
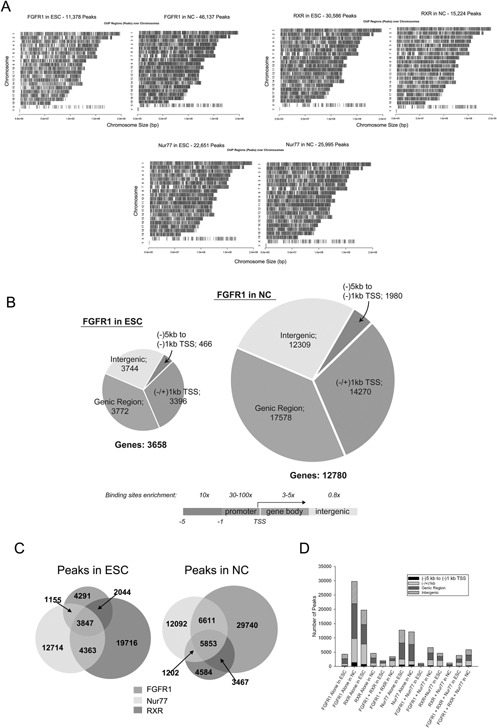
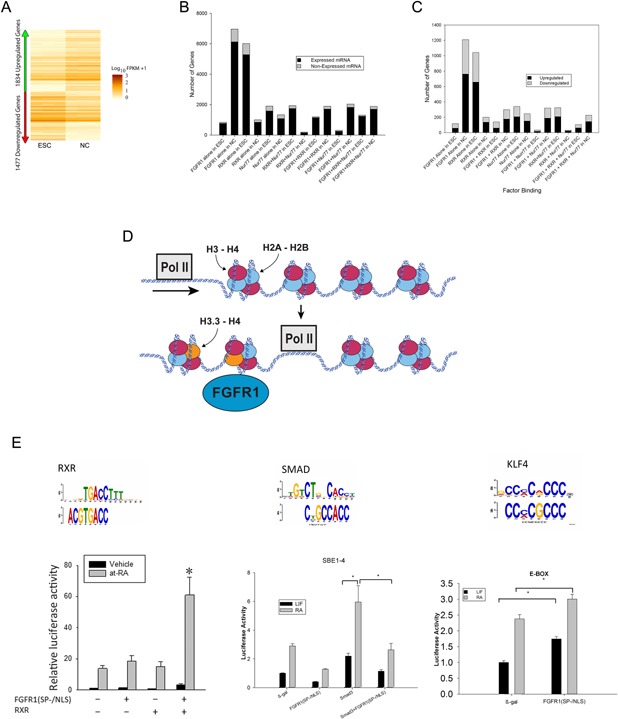
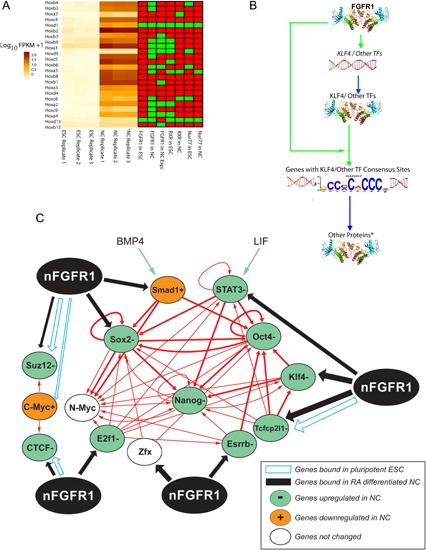
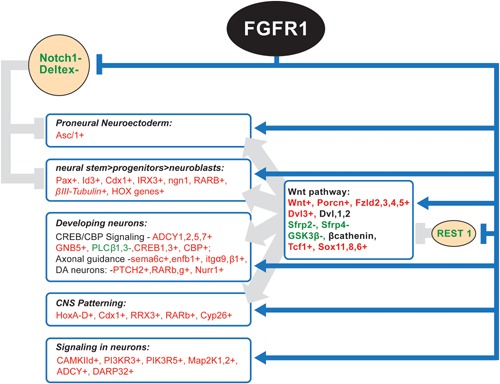
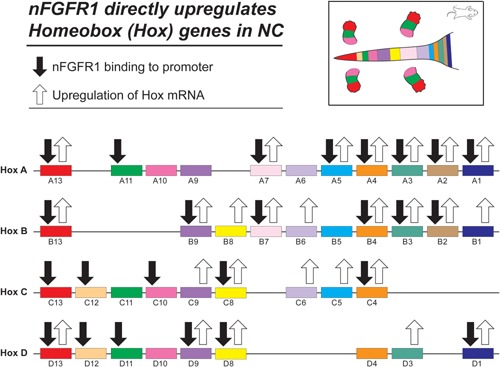
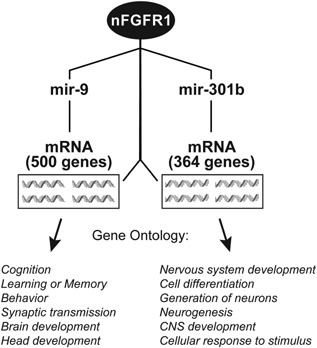
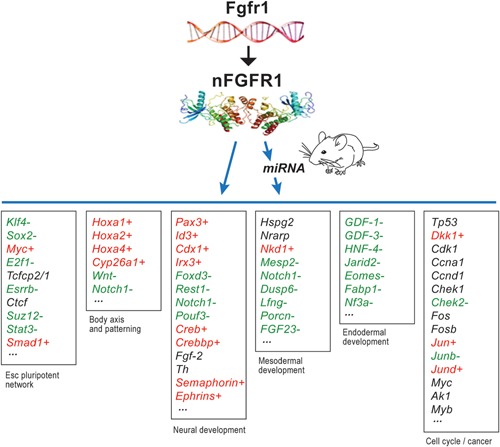
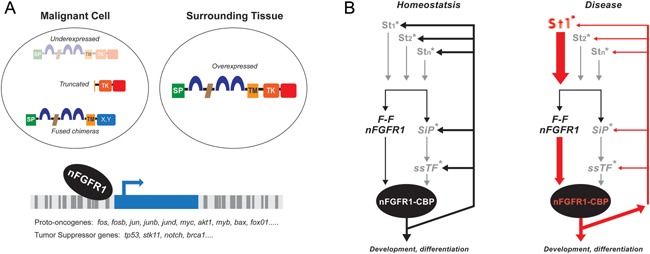
Genetic experiments have positioned the fgfr1 gene at the top of the gene hierarchy that governs gastrulation, as well as the subsequent development of the major body axes, nervous system, muscles, and bones, by affecting downstream genes that control the cell cycle, pluripotency, and differentiation, as well as microRNAs. Studies show that this regulation is executed by a single protein, the nuclear isoform of FGFR1 (nFGFR1), which integrates signals from development-initiating factors, such as retinoic acid (RA), and operates at the interface of genomic and epigenomic information. nFGFR1 cooperates with a multitude of transcriptional factors (TFs), and targets thousands of genes encoding for mRNAs, as well as miRNAs in top ontogenic networks. nFGFR1 binds to the promoters of ancient proto-oncogenes and tumor suppressor genes, in addition to binding to metazoan morphogens that delineate body axes, and construct the nervous system, as well as mesodermal and endodermal tissues. The discovery of pan-ontogenic gene programming by integrative nuclear FGFR1 signaling (INFS) impacts our understanding of ontogeny, as well as developmental pathologies, and holds new promise for reconstructive medicine, and cancer therapy.
© 2016 The Authors. Journal of Cellular Physiology published by Wiley Periodicals, Inc.
Figures
References
-
- Alexander T, Nolte C, Krumlauf R. 2009. Hox genes and segmentation of the hindbrain and axial skeleton. Annu Rev Cell Dev Biol 25:431–456. - PubMed
-
- Ayalew M, Le‐Niculescu H, Levey DF, Jain N, Changala B, Patel SD, Winiger E, Breier A, Shekhar A, Amdur R, Koller D, Nurnberger JI, Corvin A, Geyer M, Tsuang MT, Salomon D, Schork NJ, Fanous AH, O'Donovan MC, Niculescu AB. 2012. Convergent functional genomics of schizophrenia: From comprehensive understanding to genetic risk prediction. Mol Psychiatry 17:887–905. - PMC - PubMed
-
- Baron O, Forthmann B, Lee YW, Terranova C, Ratzka A, Stachowiak EK, Grothe C, Claus P, Stachowiak MK. 2012. Cooperation of nuclear fibroblast growth factor receptor 1 and Nurr1 offers new interactive mechanism in postmitotic development of mesencephalic dopaminergic neurons. J Biol Chem 287:19827–19840. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
