

Pathway Analysis: State of the Art
- PMID: 26733877
- PMCID: PMC4681784
- DOI: 10.3389/fphys.2015.00383
Pathway Analysis: State of the Art
Abstract
Pathway analysis is a set of widely used tools for research in life sciences intended to give meaning to high-throughput biological data. The methodology of these tools settles in the gathering and usage of knowledge that comprise biomolecular functioning, coupled with statistical testing and other algorithms. Despite their wide employment, pathway analysis foundations and overall background may not be fully understood, leading to misinterpretation of analysis results. This review attempts to comprise the fundamental knowledge to take into consideration when using pathway analysis as a hypothesis generation tool. We discuss the key elements that are part of these methodologies, their capabilities and current deficiencies. We also present an overview of current and all-time popular methods, highlighting different classes across them. In doing so, we show the exploding diversity of methods that pathway analysis encompasses, point out commonly overlooked caveats, and direct attention to a potential new class of methods that attempt to zoom the analysis scope to the sample scale.
Keywords: bioinformatics; functional class scoring; high-throughput biological data; over representation; pathway analysis; pathway-topology; systems biology.
Figures
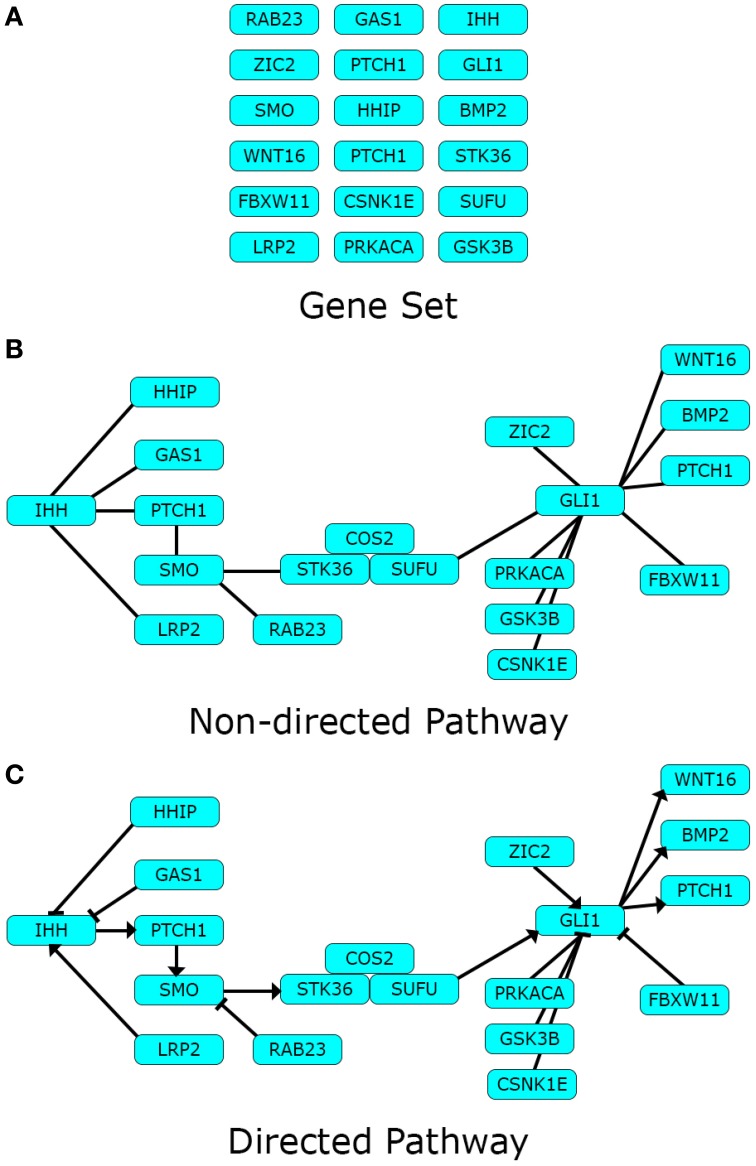


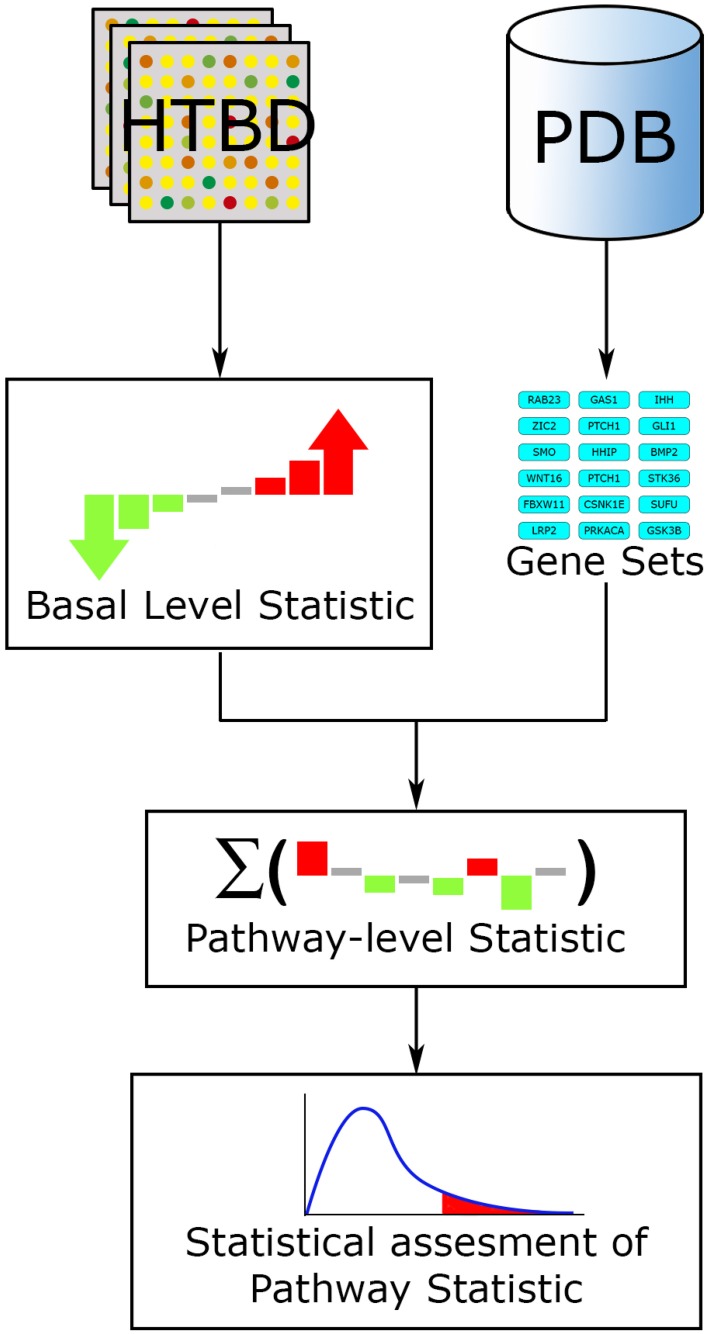
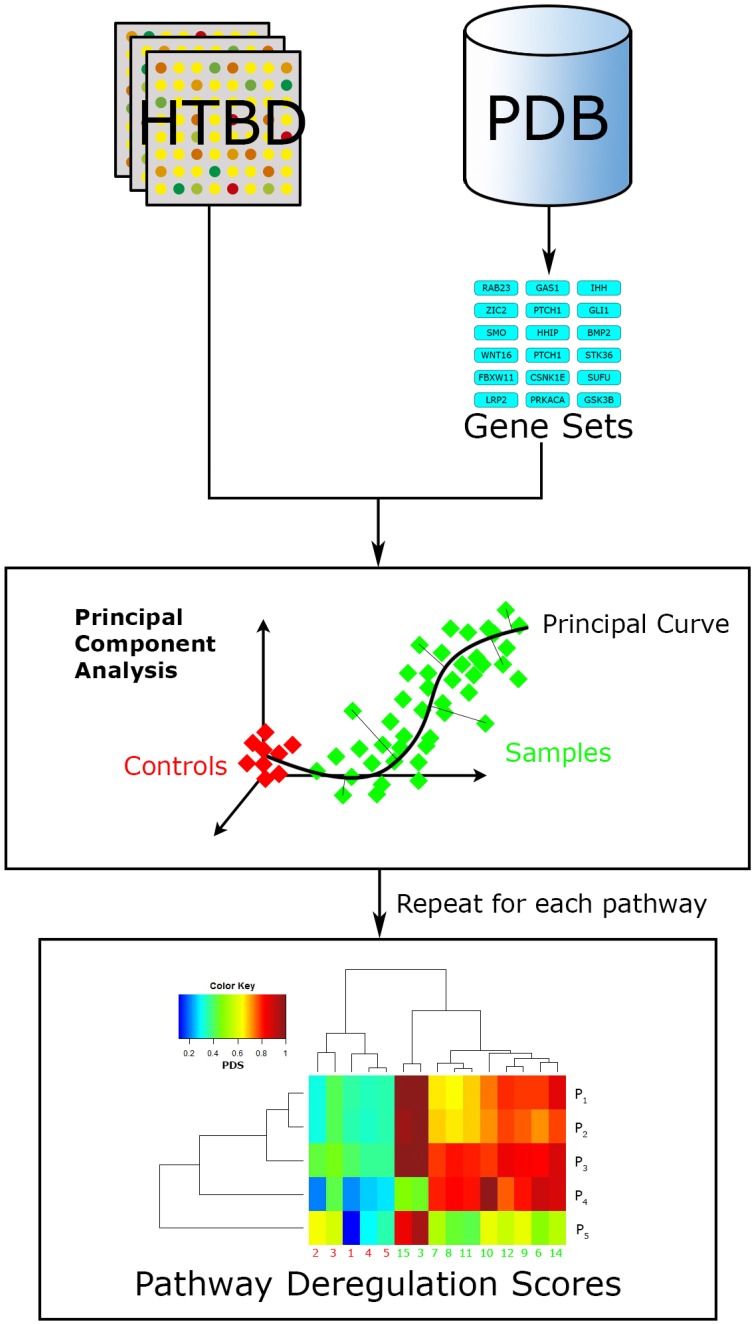
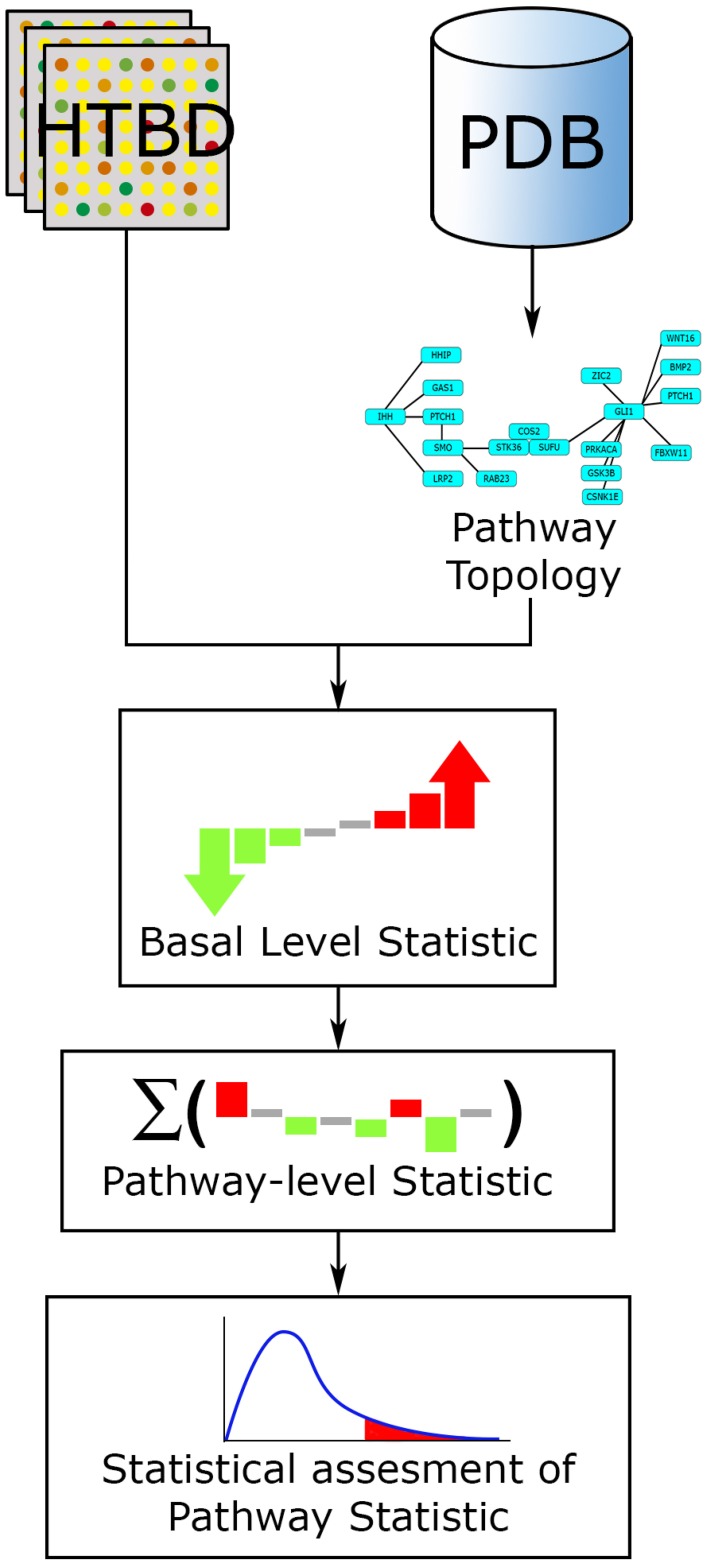
References
-
- Amaral L. A., Ottino J. M. (2004). Complex networks. Eur. Phys. J. B Condens. Matter Complex Syst. 38, 147–162. 10.1140/epjb/e2004-00110-5 - DOI
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources