

Prospects for gene-engineered T cell immunotherapy for solid cancers
- PMID: 26735408
- PMCID: PMC6295670
- DOI: 10.1038/nm.4015
Prospects for gene-engineered T cell immunotherapy for solid cancers
Abstract
Adoptive transfer of receptor-engineered T cells has produced impressive results in treating patients with B cell leukemias and lymphomas. This success has captured public imagination and driven academic and industrial researchers to develop similar 'off-the-shelf' receptors targeting shared antigens on epithelial cancers, the leading cause of cancer-related deaths. However, the successful treatment of large numbers of people with solid cancers using this strategy is unlikely to be straightforward. Receptor-engineered T cells have the potential to cause lethal toxicity from on-target recognition of normal tissues, and there is a paucity of truly tumor-specific antigens shared across tumor types. Here we offer our perspective on how expanding the use of genetically redirected T cells to treat the majority of patients with solid cancers will require major technical, manufacturing and regulatory innovations centered around the development of autologous gene therapies targeting private somatic mutations.
Conflict of interest statement
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the
Figures
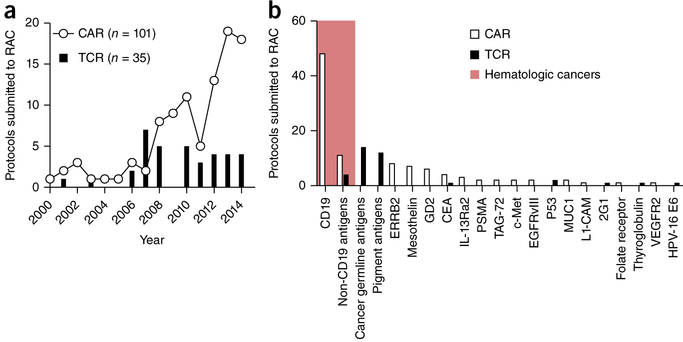
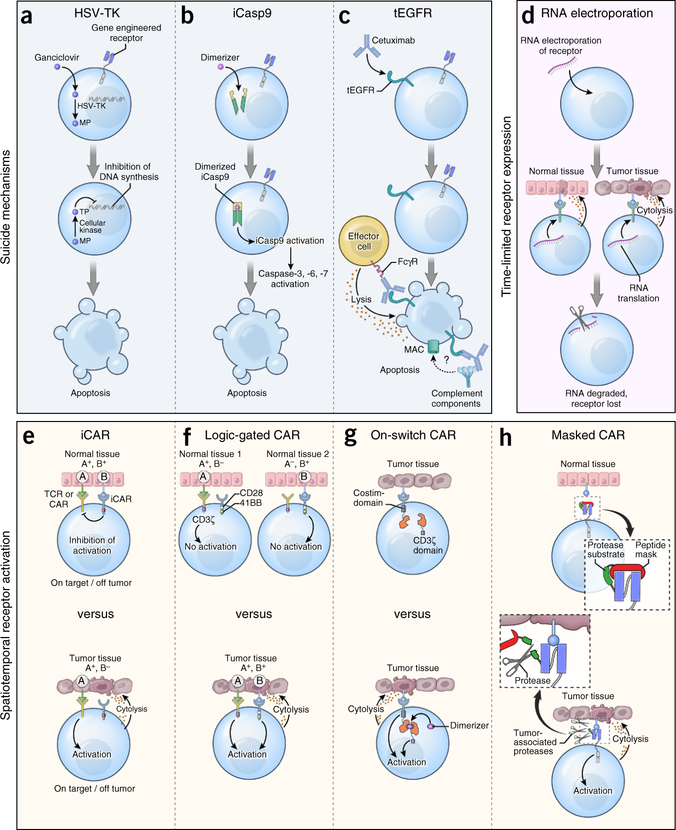
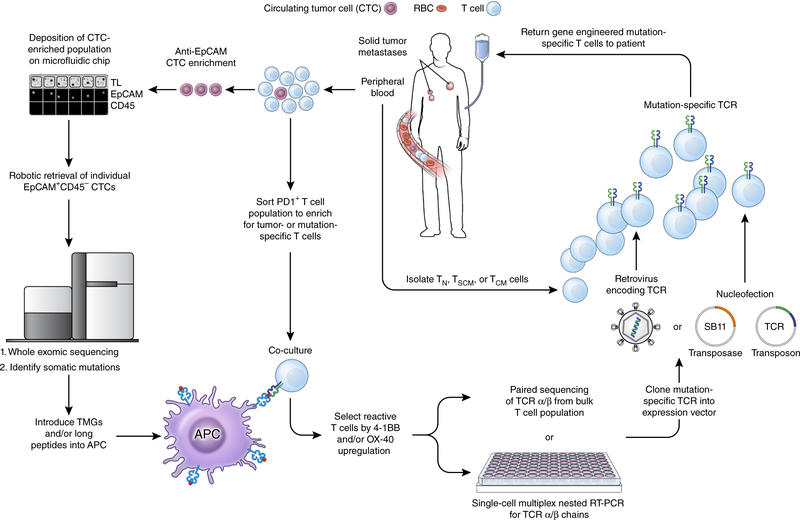
References
-
- Powles T et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 515, 558–562 (2014). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
