

126 novel mutations in Italian patients with neurofibromatosis type 1
- PMID: 26740943
- PMCID: PMC4694136
- DOI: 10.1002/mgg3.161
126 novel mutations in Italian patients with neurofibromatosis type 1
Abstract
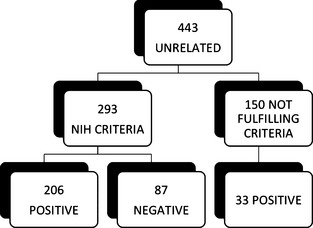
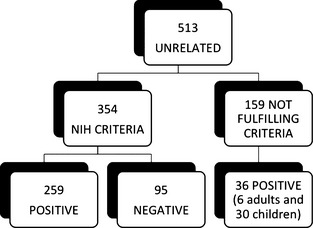
Genetic analysis of Neurofibromatosis type 1 (NF1) may facilitate the identification of patients in early phases of the disease. Here, we present an overview of our diagnostic research spanning the last 11 years, with a focus on the description of 225 NF1 mutations, 126 of which are novel, found in a series of 607 patients (513 unrelated) in Italy. Between 2003 and 2013, 443 unrelated patients were profiled by denaturing high pressure liquid chromatography (DHPLC) analysis of 60 amplicons derived from genomic NF1 DNA and subsequent sequencing of heterozygotic PCR products. In addition, a subset of patients was studied by multiplex ligation-dependent probe amplification (MLPA) to identify any duplications, large deletions or microdeletions present at the locus. Over the last year, 70 unrelated patients were investigated by MLPA and sequencing of 22 amplicons spanning the entire NF1 cDNA. Mutations were found in 70% of the 293 patients studied by DHPLC, thereby fulfilling the NIH criterion for the clinical diagnosis of NF1 (detection rate: 70%); furthermore, 87% of the patients studied by RNA sequencing were genetically characterized. Mutations were also found in 36 of the 159 patients not fulfilling the NIH clinical criteria. We confirmed a higher incidence of intellectual disability in patients harboring microdeletion type 1 and observed a correlation between a mild phenotype and the small deletion c.2970_2972delAAT or the missense alteration in amino acid residue 1809 (p.Arg1809Cys). These data support the use of RNA-based methods for genetic analysis and provide novel information for improving the management of symptoms in oligosymptomatic patients.
Keywords: Diagnostic criteria; mutation database; neurofibromatosis type 1 (NF1); novel mutations.
Figures




References
-
- Bengesser, K. , Cooper D. N., Steinmann K., Kluwe L., Chuzhanova N. A., Wimmer K., et al. 2010. A novel third type of recurrent NF1 microdeletion mediated by nonallelic homologous recombination between LRRC37B‐containing low‐copy repeats in 17q11.2. Hum. Mutat. 31:742–751. - PubMed
-
- Brems, H. , and Legius E.. 2013. Legius syndrome, an update. Molecular pathology of mutations in SPRED1. Keio J. Med. 62:107–112. - PubMed
-
- Brems, H. , Chmara M., Sahbatou M., Denayer E., Taniguchi K., Kato R., et al. 2007. Germline loss‐of‐function mutations in SPRED1 cause a neurofibromatosis 1‐like phenotype. Nat. Genet. 39:1120–1126. - PubMed
-
- Burkitt Wright, E. M. , Sach E., Sharif S., Quarrell O., Carroll T., Whitehouse R. W., et al. 2013. Can the diagnosis of NF1 be excluded clinically? A lack of pigmentary findings in families with spinal neurofibromatosis demonstrates a limitation of clinical diagnosis. J. Med. Genet. 50:606–613. - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
