

Testicular dysgenesis/regression without campomelic dysplasia in patients carrying missense mutations and upstream deletion of SOX9
- PMID: 26740947
- PMCID: PMC4694128
- DOI: 10.1002/mgg3.165
Testicular dysgenesis/regression without campomelic dysplasia in patients carrying missense mutations and upstream deletion of SOX9
Abstract
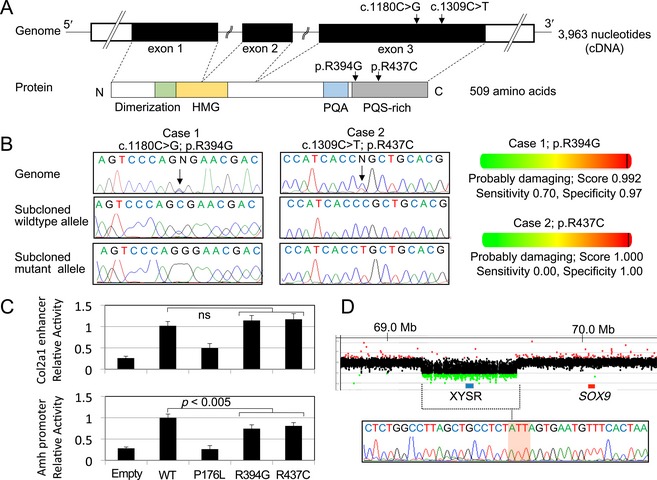
SOX9 haploinsufficiency underlies campomelic dysplasia (CD) with or without testicular dysgenesis. Current understanding of the phenotypic variability and mutation spectrum of SOX9 abnormalities remains fragmentary. Here, we report three patients with hitherto unreported SOX9 abnormalities. These patients were identified through molecular analysis of 33 patients with 46,XY disorders of sex development (DSD). Patients 1-3 manifested testicular dysgenesis or regression without CD. Patients 1 and 2 carried probable damaging mutations p.Arg394Gly and p.Arg437Cys, respectively, in the SOX9 C-terminal domain but not in other known 46,XY DSD causative genes. These substitutions were absent from ~120,000 alleles in the exome database. These mutations retained normal transactivating activity for the Col2a1 enhancer, but showed impaired activity for the Amh promoter. Patient 3 harbored a maternally inherited ~491 kb SOX9 upstream deletion that encompassed the known 32.5 kb XY sex reversal region. Breakpoints of the deletion resided within nonrepeat sequences and were accompanied by a short-nucleotide insertion. The results imply that testicular dysgenesis and regression without skeletal dysplasia may be rare manifestations of SOX9 abnormalities. Furthermore, our data broaden pathogenic SOX9 abnormalities to include C-terminal missense substitutions which lead to target-gene-specific protein dysfunction, and enhancer-containing upstream microdeletions mediated by nonhomologous end-joining.
Keywords: Campomelic dysplasia; deletion; enhancer; mutation; testis.
Figures
Similar articles
-
Copy number variation of two separate regulatory regions upstream of SOX9 causes isolated 46,XY or 46,XX disorder of sex development.J Med Genet. 2015 Apr;52(4):240-7. doi: 10.1136/jmedgenet-2014-102864. Epub 2015 Jan 20. J Med Genet. 2015. PMID: 25604083
-
The clinical impact of chromosomal rearrangements with breakpoints upstream of the SOX9 gene: two novel de novo balanced translocations associated with acampomelic campomelic dysplasia.BMC Med Genet. 2013 May 7;14:50. doi: 10.1186/1471-2350-14-50. BMC Med Genet. 2013. PMID: 23648064 Free PMC article.
-
Mutational analysis of the SOX9 gene in campomelic dysplasia and autosomal sex reversal: lack of genotype/phenotype correlations.Hum Mol Genet. 1997 Jan;6(1):91-8. doi: 10.1093/hmg/6.1.91. Hum Mol Genet. 1997. PMID: 9002675
-
The molecular action of testis-determining factors SRY and SOX9.Novartis Found Symp. 2002;244:57-66; discussion 66-7, 79-85, 253-7. Novartis Found Symp. 2002. PMID: 11990798 Review.
-
SOX9 in organogenesis: shared and unique transcriptional functions.Cell Mol Life Sci. 2022 Sep 17;79(10):522. doi: 10.1007/s00018-022-04543-4. Cell Mol Life Sci. 2022. PMID: 36114905 Free PMC article. Review.
Cited by
-
De Novo SOX4 Variants Cause a Neurodevelopmental Disease Associated with Mild Dysmorphism.Am J Hum Genet. 2019 Feb 7;104(2):246-259. doi: 10.1016/j.ajhg.2018.12.014. Epub 2019 Jan 17. Am J Hum Genet. 2019. PMID: 30661772 Free PMC article.
-
Diverse Regulation but Conserved Function: SOX9 in Vertebrate Sex Determination.Genes (Basel). 2021 Mar 26;12(4):486. doi: 10.3390/genes12040486. Genes (Basel). 2021. PMID: 33810596 Free PMC article. Review.
-
The SOXE transcription factors-SOX8, SOX9 and SOX10-share a bi-partite transactivation mechanism.Nucleic Acids Res. 2019 Jul 26;47(13):6917-6931. doi: 10.1093/nar/gkz523. Nucleic Acids Res. 2019. PMID: 31194875 Free PMC article.
-
Case report: A de novo Non-sense SOX9 mutation (p.Q417*) located in transactivation domain is Responsible for Campomelic Dysplasia.Front Pediatr. 2023 Jan 18;10:1089194. doi: 10.3389/fped.2022.1089194. eCollection 2022. Front Pediatr. 2023. PMID: 36741086 Free PMC article.
-
Next generation sequencing and array-based comparative genomic hybridization for molecular diagnosis of pediatric endocrine disorders.Ann Pediatr Endocrinol Metab. 2017 Jun;22(2):90-94. doi: 10.6065/apem.2017.22.2.90. Epub 2017 Jun 28. Ann Pediatr Endocrinol Metab. 2017. PMID: 28690986 Free PMC article. Review.
References
-
- Bagheri‐Fam, S. , Barrionuevo F., Dohrmann U., Gunther T., Schule R., Kemler R., et al. 2006. Long‐range upstream and downstream enhancers control distinct subsets of the complex spatiotemporal Sox9 expression pattern. Dev. Biol. 291:382–397. - PubMed
-
- Barrionuevo, F. , Georg I., Scherthan H., Lecureuil C., Guillou F., Wegner M., et al. 2009. Testis cord differentiation after the sex determination stage is independent of Sox9 but fails in the combined absence of Sox9 and Sox8. Dev. Biol. 327:301–312. - PubMed
-
- Bernard, P. , Tang P., Liu S., Dewing P., Harley V. R., and E. Vilain . 2003. Dimerization of SOX9 is required for chondrogenesis, but not for sex determination. Hum. Mol. Genet. 12:1755–1765. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
