

Comparative Study
doi: 10.1007/s00439-015-1631-9.
Epub 2016 Jan 7.
Clinical sequencing: is WGS the better WES?
Affiliations
- PMID: 26742503
- PMCID: PMC4757617
- DOI: 10.1007/s00439-015-1631-9
Item in Clipboard
Comparative Study
Clinical sequencing: is WGS the better WES?
Hum Genet.
2016 Mar.
Abstract
Current clinical next-generation sequencing is done by using gene panels and exome analysis, both of which involve selective capturing of target regions. However, capturing has limitations in sufficiently covering coding exons, especially GC-rich regions. We compared whole exome sequencing (WES) with the most recent PCR-free whole genome sequencing (WGS), showing that only the latter is able to provide hitherto unprecedented complete coverage of the coding region of the genome. Thus, from a clinical/technical point of view, WGS is the better WES so that capturing is no longer necessary for the most comprehensive genomic testing of Mendelian disorders.
Figures
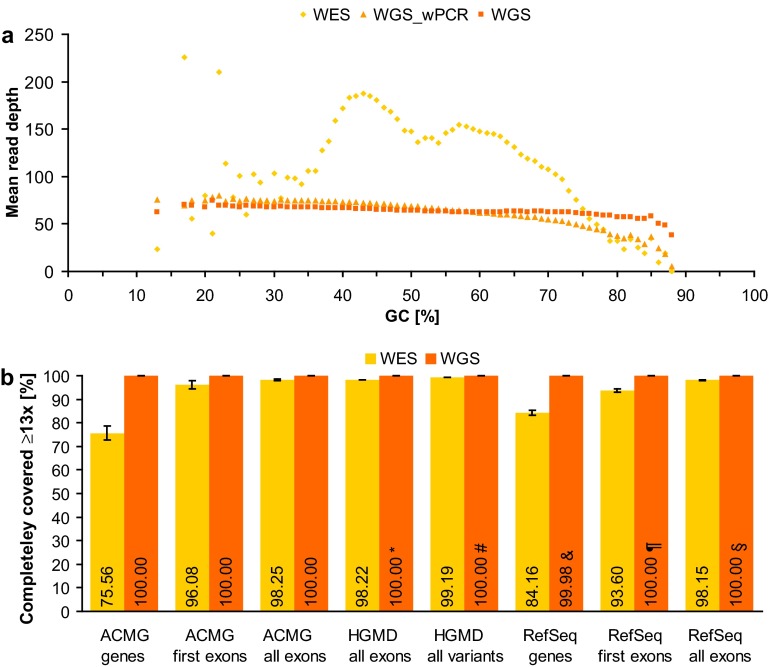
Performance comparison of WES and WGS. a Mean read depth of RefSeq coding exons per GC content shown for WES as well as for WGS with (WGS_wPCR) and without (WGS) PCR as means of five samples each. b Percentage of completely covered (i.e. ≥13 reads at each nucleotide position) genes, exons, and variants in WES and WGS without PCR as means of five samples each (error bars indicate 95 % confidence intervals). In the case of genes recommended for reporting by the ACMG (ACMG genes, n = 54) and of genes of the RefSeq database (RefSeq genes, n = 16,896), the set of all coding exons (ACMG all exons, n = 1152; RefSeq all exons, n = 177,084) and the set of start-codon-containing exons (first exons) were examined. The set of RefSeq exons harboring at least one disease-causing mutation (DM) listed in HGMD (HGMD all exons, n = 22,303) and the set of all coding and non-coding DMs (HGMD all variants, n = 106,819) were also analyzed. Note that 100.00 % implies a deviation of at most 0.005 %: *two exons were partially covered with 12 reads; #one intronic mutation was covered with 12 reads; &12 genes were partially covered with 7–12 reads; ¶three exons were partially covered with 10–12 reads; §12 exons were partially covered with 7–12 reads
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
