

Elevated glucose and oligomeric β-amyloid disrupt synapses via a common pathway of aberrant protein S-nitrosylation
- PMID: 26743041
- PMCID: PMC4729876
- DOI: 10.1038/ncomms10242
Elevated glucose and oligomeric β-amyloid disrupt synapses via a common pathway of aberrant protein S-nitrosylation
Abstract
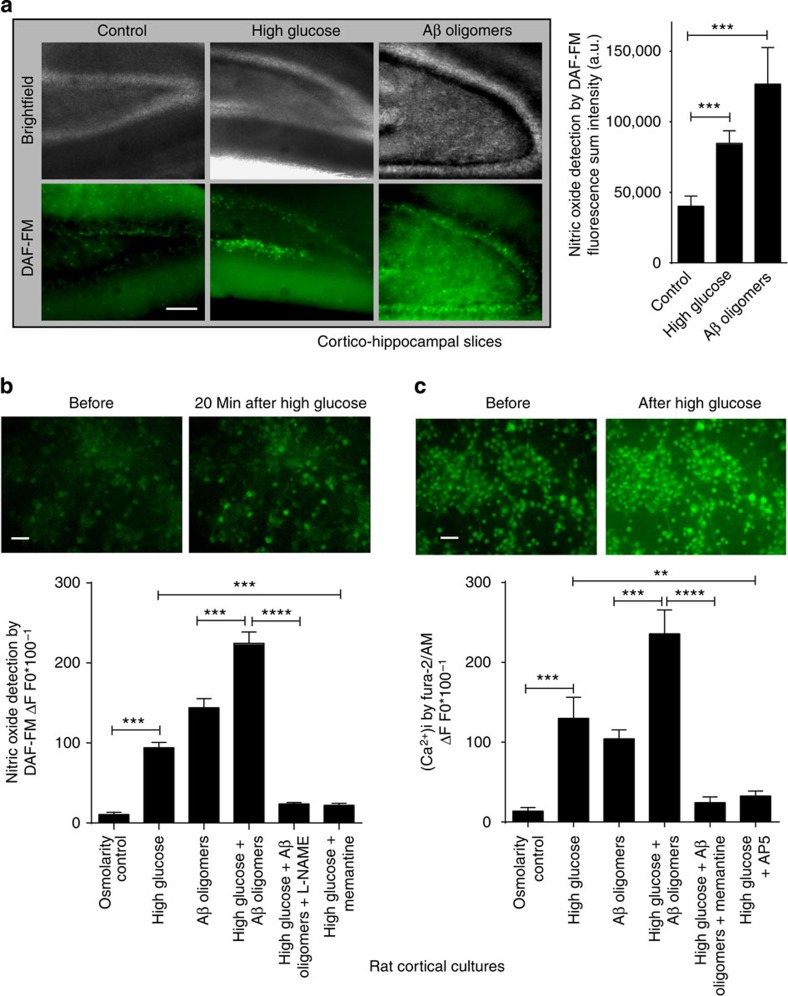
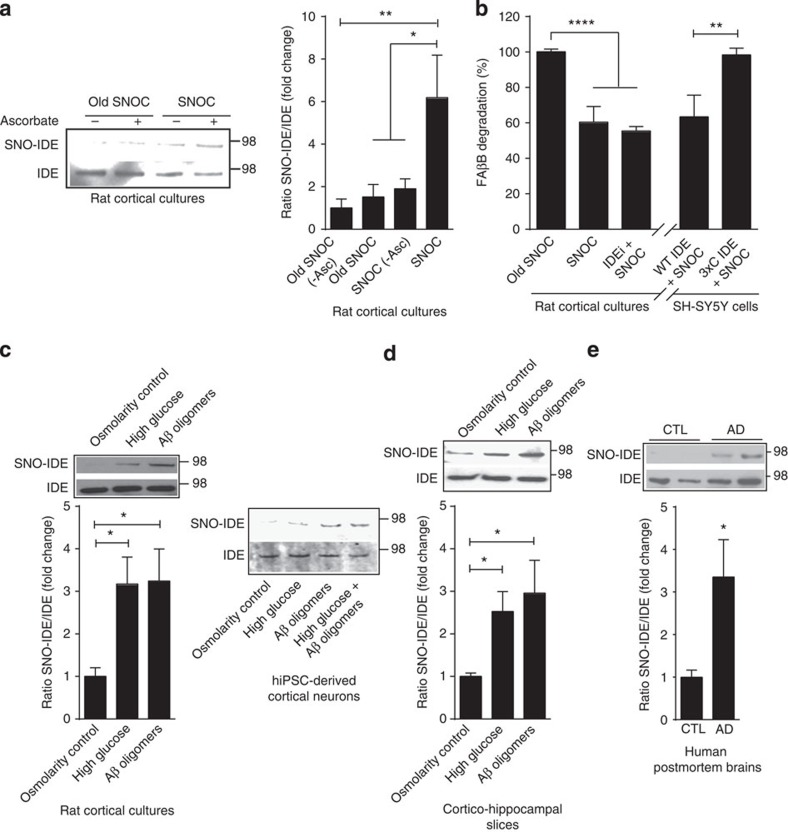
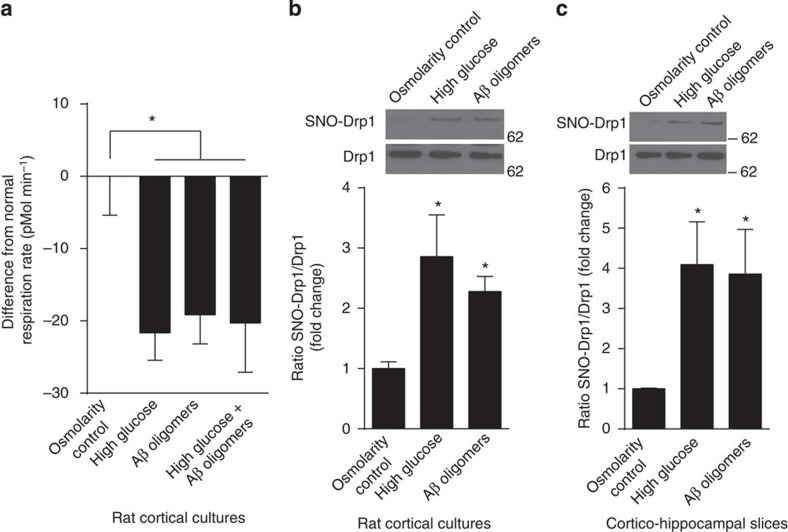
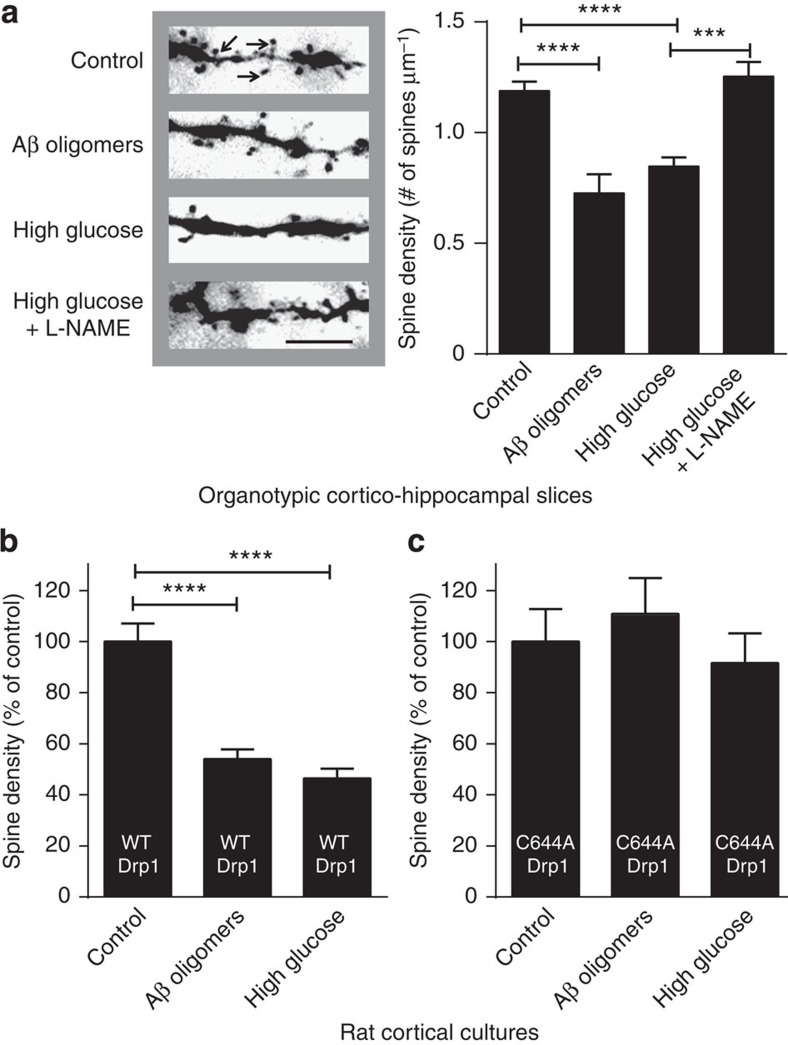
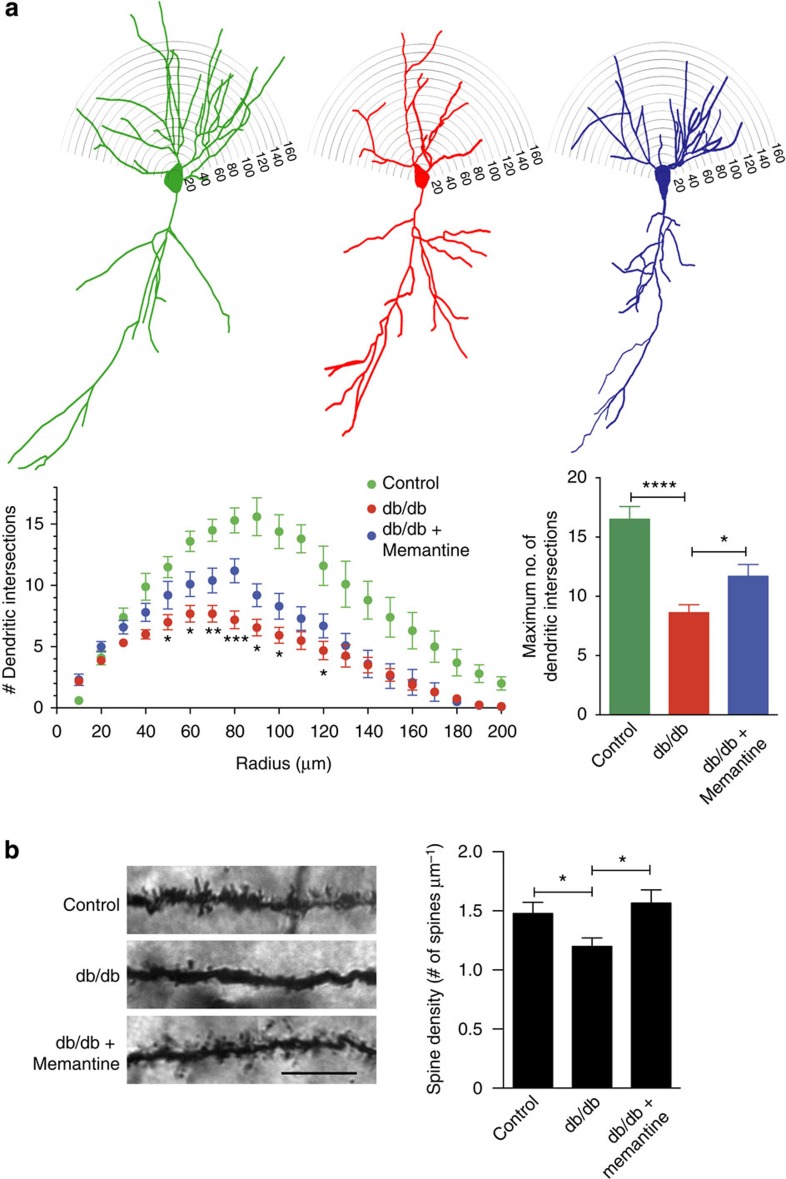
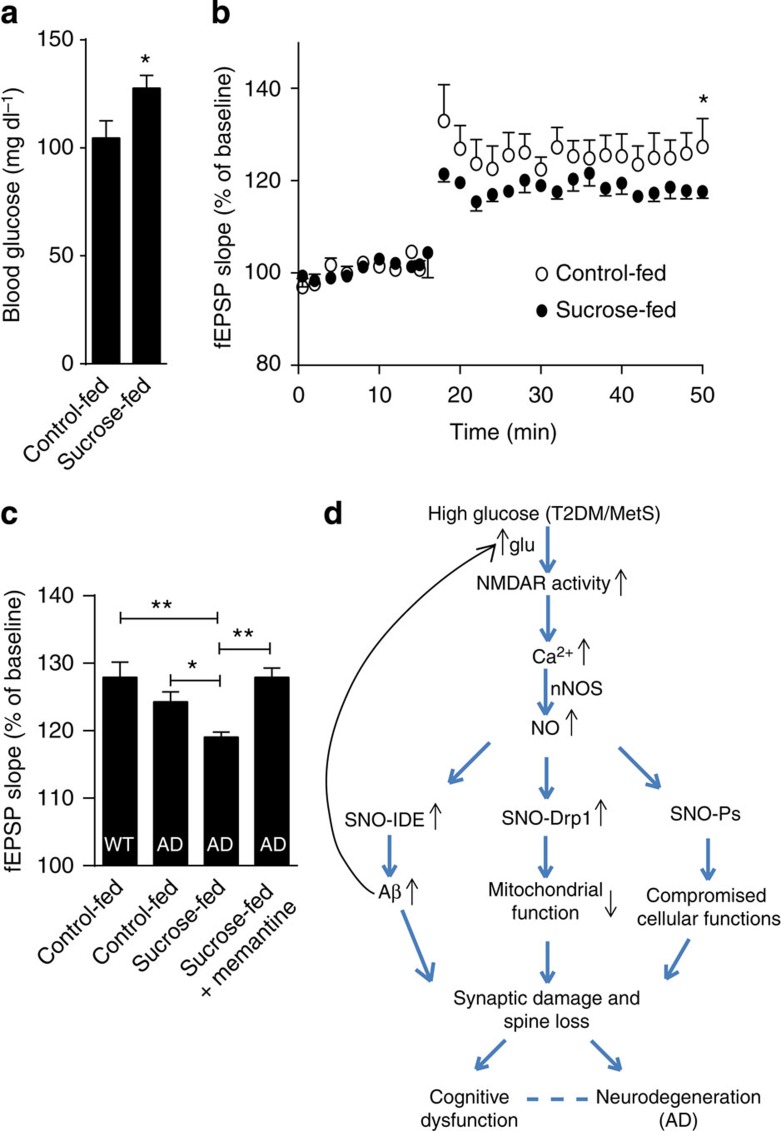
Metabolic syndrome (MetS) and Type 2 diabetes mellitus (T2DM) increase risk for Alzheimer's disease (AD). The molecular mechanism for this association remains poorly defined. Here we report in human and rodent tissues that elevated glucose, as found in MetS/T2DM, and oligomeric β-amyloid (Aβ) peptide, thought to be a key mediator of AD, coordinately increase neuronal Ca(2+) and nitric oxide (NO) in an NMDA receptor-dependent manner. The increase in NO results in S-nitrosylation of insulin-degrading enzyme (IDE) and dynamin-related protein 1 (Drp1), thus inhibiting insulin and Aβ catabolism as well as hyperactivating mitochondrial fission machinery. Consequent elevation in Aβ levels and compromise in mitochondrial bioenergetics result in dysfunctional synaptic plasticity and synapse loss in cortical and hippocampal neurons. The NMDA receptor antagonist memantine attenuates these effects. Our studies show that redox-mediated posttranslational modification of brain proteins link Aβ and hyperglycaemia to cognitive dysfunction in MetS/T2DM and AD.
Conflict of interest statement
S.A.L. is the named inventor on worldwide patents for memantine (Namenda®) for the treatment of neurodegenerative diseases. Following Harvard University guidelines, he participates in a royalty sharing agreement with his former institution Harvard Medical School/Boston Children's Hospital for the licensing of these patents to Forest Laboratories (now Actavis, Inc.). He is also a Scientific Founder of Adamas Pharmaceuticals, Inc., which has agreements with Forest Laboratories/Actavis for the co-development and marketing of long-lasting formulations of memantine. The remaining authors declare competing interests.
Figures
References
-
- Biessels G. J., Staekenborg S., Brunner E., Brayne C. & Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 5, 64–74 (2006) . - PubMed
-
- Ramos-Rodriguez J. J. et al. Differential central pathology and cognitive impairment in pre-diabetic and diabetic mice. Psychoneuroendocrinology 38, 2462–2475 (2013) . - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
