

Fast and accurate branch lengths estimation for phylogenomic trees
- PMID: 26744021
- PMCID: PMC4705742
- DOI: 10.1186/s12859-015-0821-8
Fast and accurate branch lengths estimation for phylogenomic trees
Abstract
Background: Branch lengths are an important attribute of phylogenetic trees, providing essential information for many studies in evolutionary biology. Yet, part of the current methodology to reconstruct a phylogeny from genomic information - namely supertree methods - focuses on the topology or structure of the phylogenetic tree, rather than the evolutionary divergences associated to it. Moreover, accurate methods to estimate branch lengths - typically based on probabilistic analysis of a concatenated alignment - are limited by large demands in memory and computing time, and may become impractical when the data sets are too large.
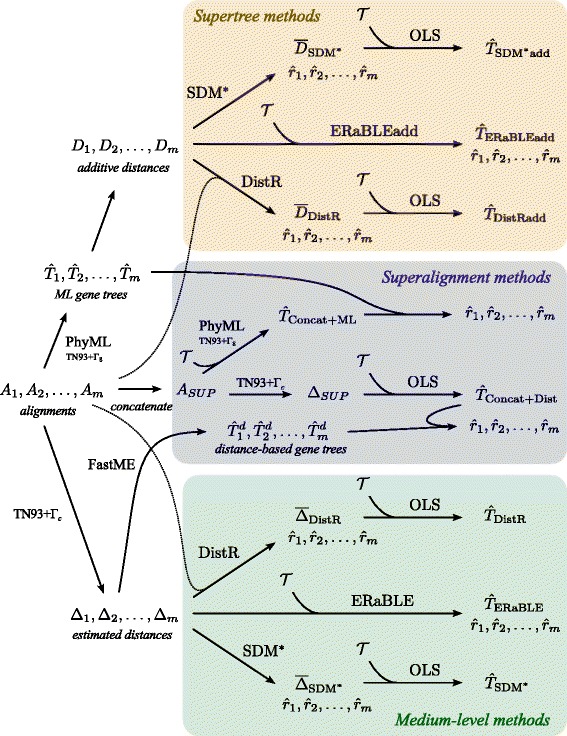
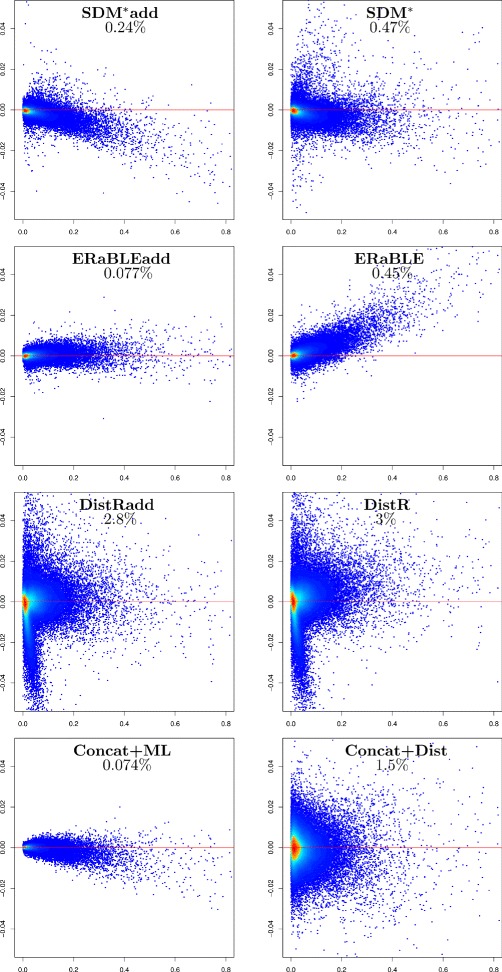
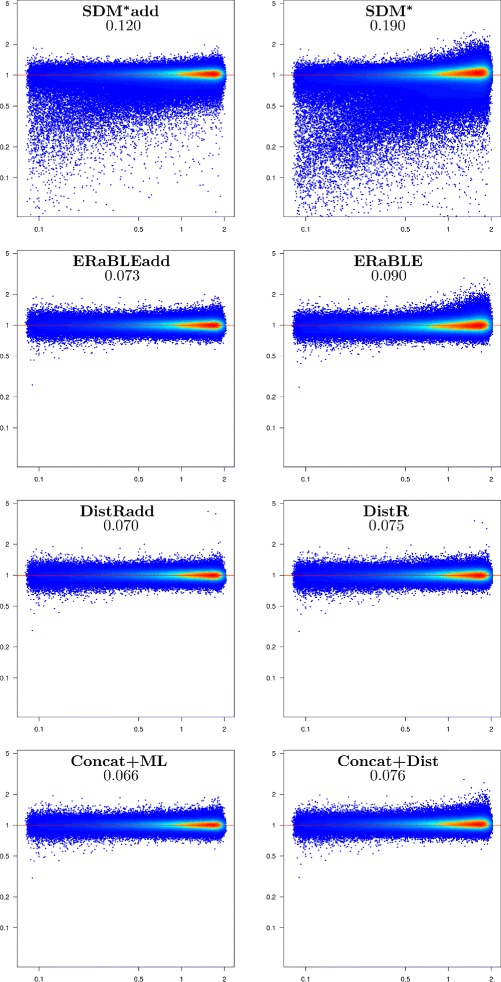
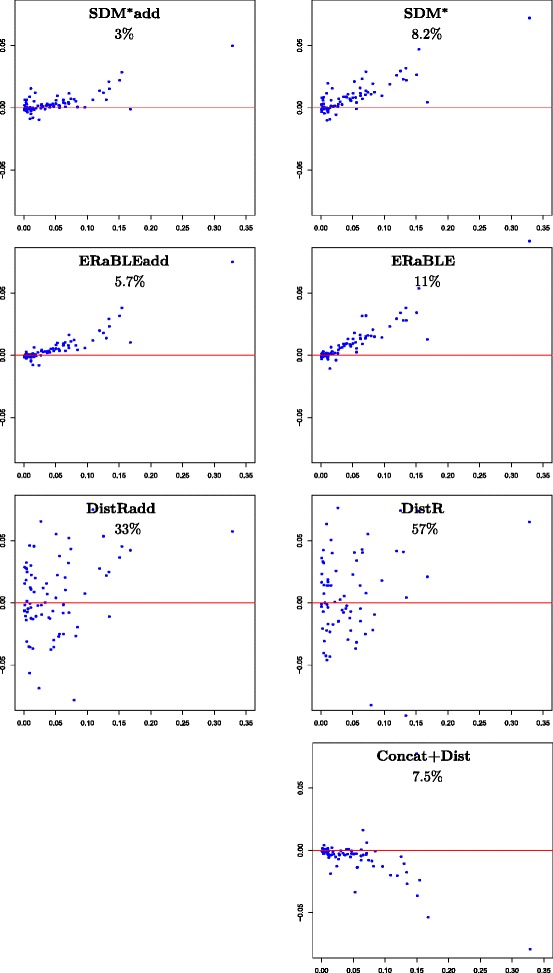
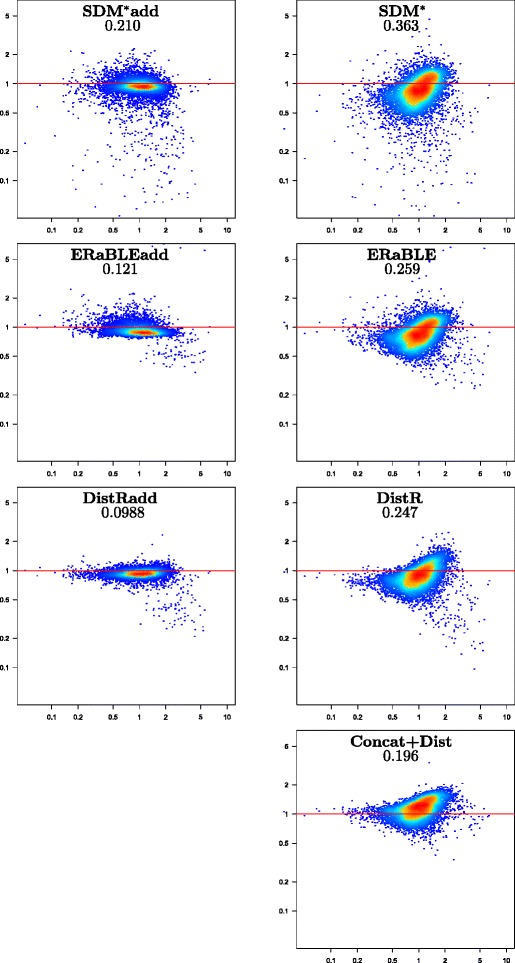
Results: Here, we present a novel phylogenomic distance-based method, named ERaBLE (Evolutionary Rates and Branch Length Estimation), to estimate the branch lengths of a given reference topology, and the relative evolutionary rates of the genes employed in the analysis. ERaBLE uses as input data a potentially very large collection of distance matrices, where each matrix is obtained from a different genomic region - either directly from its sequence alignment, or indirectly from a gene tree inferred from the alignment. Our experiments show that ERaBLE is very fast and fairly accurate when compared to other possible approaches for the same tasks. Specifically, it efficiently and accurately deals with large data sets, such as the OrthoMaM v8 database, composed of 6,953 exons from up to 40 mammals.
Conclusions: ERaBLE may be used as a complement to supertree methods - or it may provide an efficient alternative to maximum likelihood analysis of concatenated alignments - to estimate branch lengths from phylogenomic data sets.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
