

Mutations in AP3D1 associated with immunodeficiency and seizures define a new type of Hermansky-Pudlak syndrome
- PMID: 26744459
- PMCID: PMC7611501
- DOI: 10.1182/blood-2015-09-671636
Mutations in AP3D1 associated with immunodeficiency and seizures define a new type of Hermansky-Pudlak syndrome
Abstract
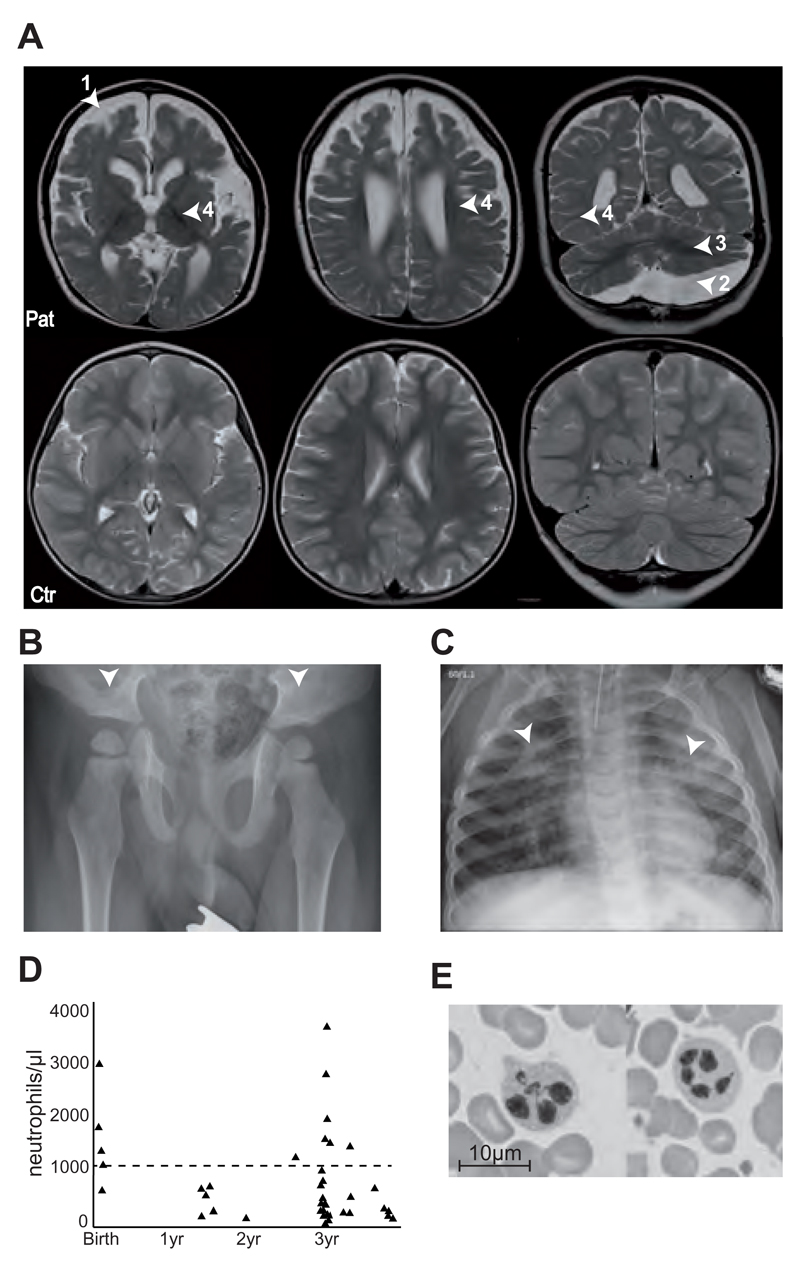
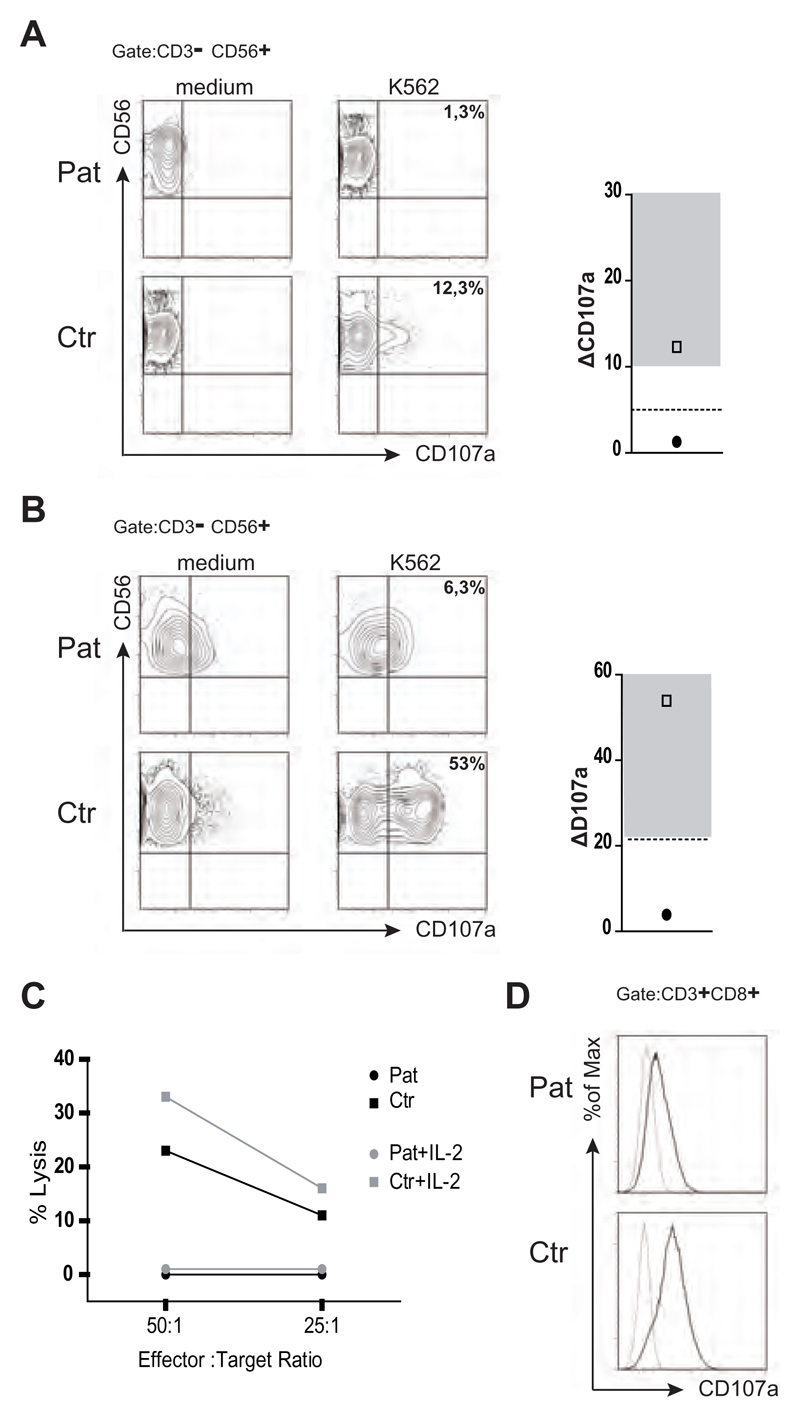
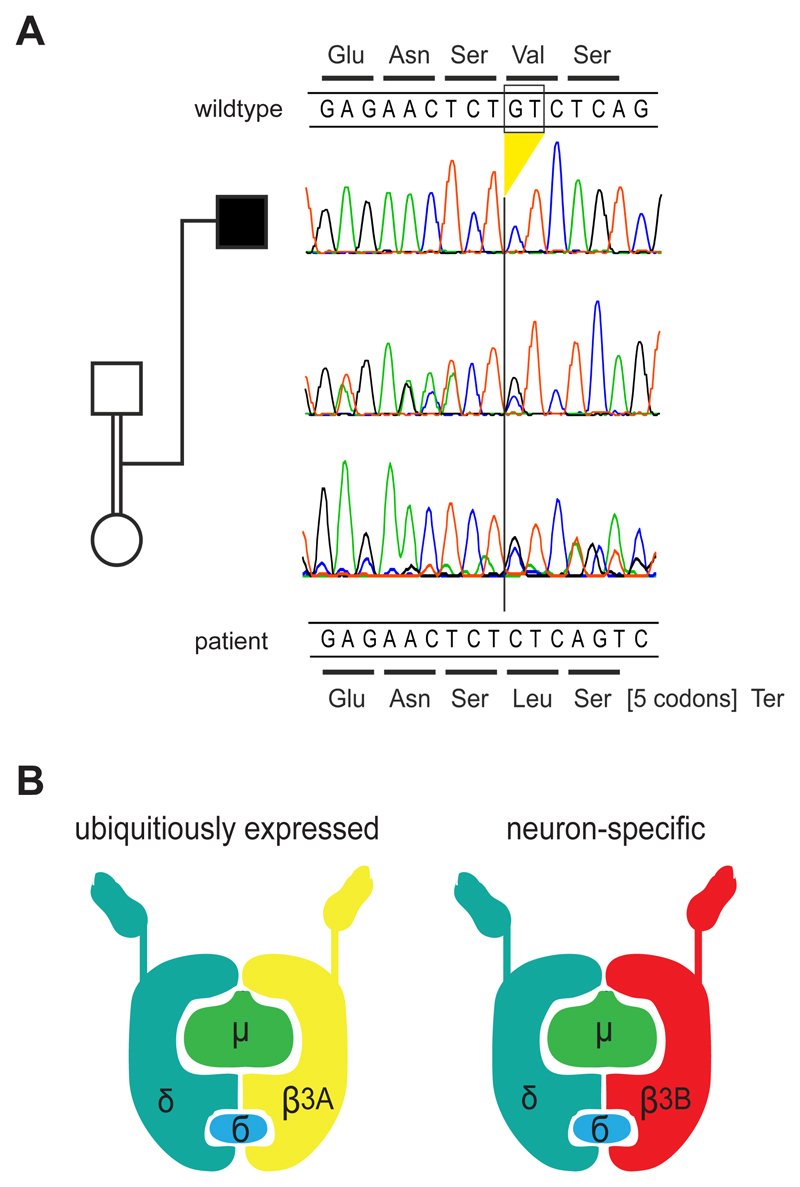
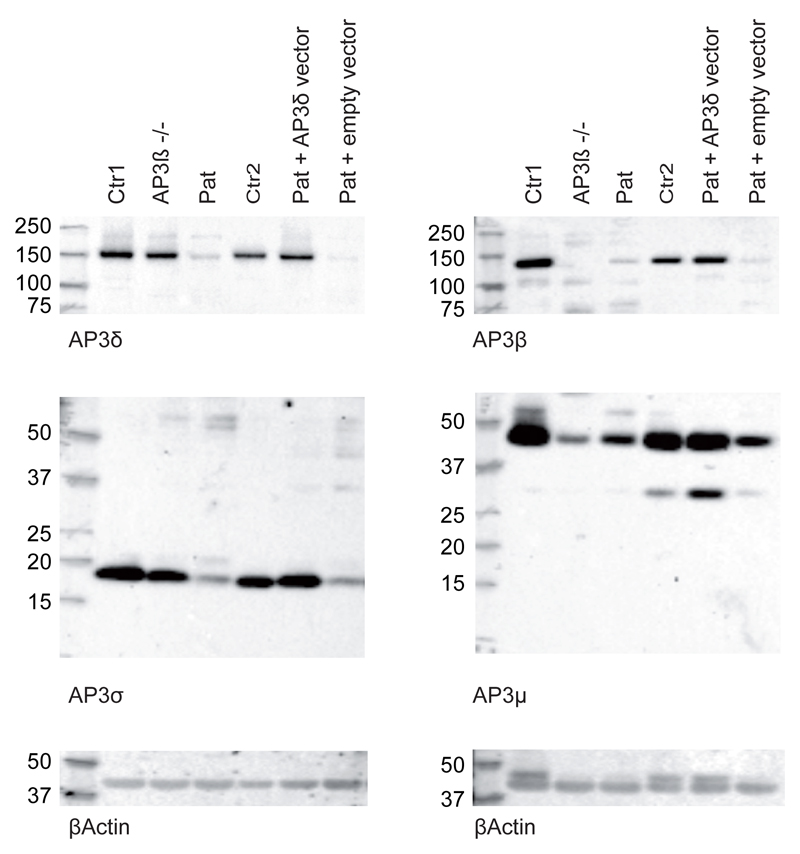
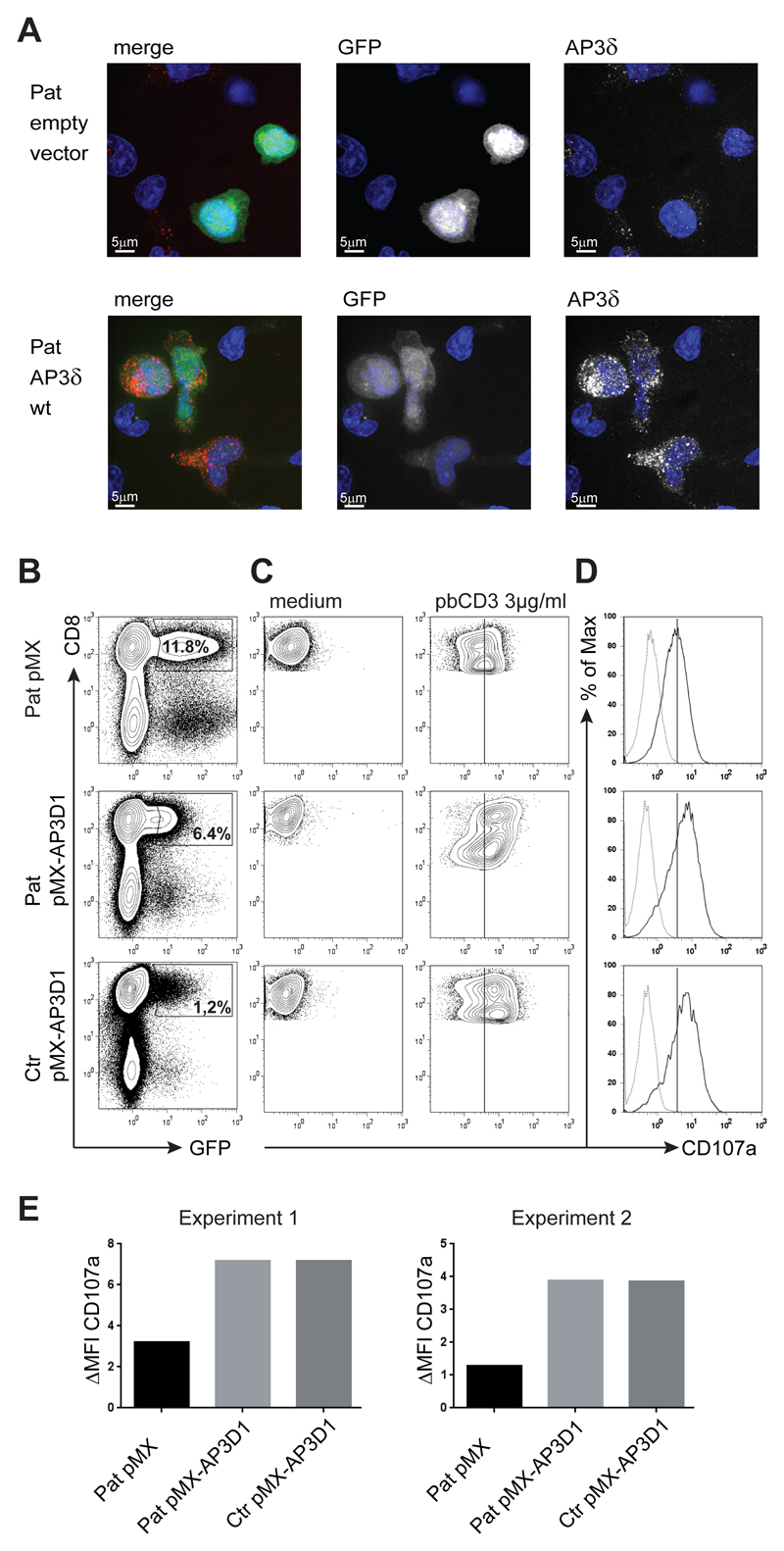
Genetic disorders affecting biogenesis and transport of lysosome-related organelles are heterogeneous diseases frequently associated with albinism. We studied a patient with albinism, neutropenia, immunodeficiency, neurodevelopmental delay, generalized seizures, and impaired hearing but with no mutation in genes so far associated with albinism and immunodeficiency. Whole exome sequencing identified a homozygous mutation in AP3D1 that leads to destabilization of the adaptor protein 3 (AP3) complex. AP3 complex formation and the degranulation defect in patient T cells were restored by retroviral reconstitution. A previously described hypopigmented mouse mutant with an Ap3d1 null mutation (mocha strain) shares the neurologic phenotype with our patient and shows a platelet storage pool deficiency characteristic of Hermansky-Pudlak syndrome (HPS) that was not studied in our patient because of a lack of bleeding. HPS2 caused by mutations in AP3B1A leads to a highly overlapping phenotype without the neurologic symptoms. The AP3 complex exists in a ubiquitous and a neuronal form. AP3D1 codes for the AP3δ subunit of the complex, which is essential for both forms. In contrast, the AP3β3A subunit, affected in HPS2 patients, is substituted by AP3β3B in the neuron-specific heterotetramer. AP3δ deficiency thus causes a severe neurologic disorder with immunodeficiency and albinism that we propose to classify as HPS10.
© 2016 by The American Society of Hematology.
Conflict of interest statement
The authors declare they have no potential conflict of interest.
Figures
Comment in
-
A new type of syndromic albinism associated with mutations in AP3D1.Pigment Cell Melanoma Res. 2017 Jan;30(1):5-7. doi: 10.1111/pcmr.12543. Epub 2016 Nov 30. Pigment Cell Melanoma Res. 2017. PMID: 27900855 Free PMC article. No abstract available.
References
-
- Griscelli C, Durandy A, Guy-Grand D, Daguillard F, Herzog C, Prunieras M. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65(4):691–702. - PubMed
-
- Blume RS, Wolff SM. The Chediak-Higashi syndrome: studies in four patients and a review of the literature. Medicine (Baltimore) 1972;51(4):247–280. - PubMed
-
- Dell'Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell. 1999;3(1):11–21. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
