

BWM*: A Novel, Provable, Ensemble-based Dynamic Programming Algorithm for Sparse Approximations of Computational Protein Design
- PMID: 26744898
- PMCID: PMC4904165
- DOI: 10.1089/cmb.2015.0194
BWM*: A Novel, Provable, Ensemble-based Dynamic Programming Algorithm for Sparse Approximations of Computational Protein Design
Abstract
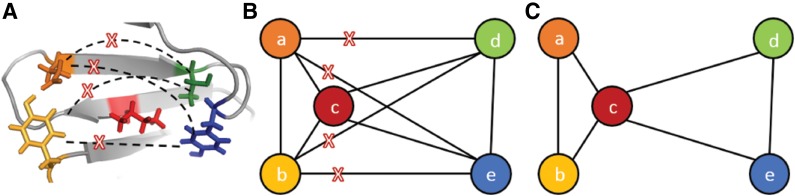
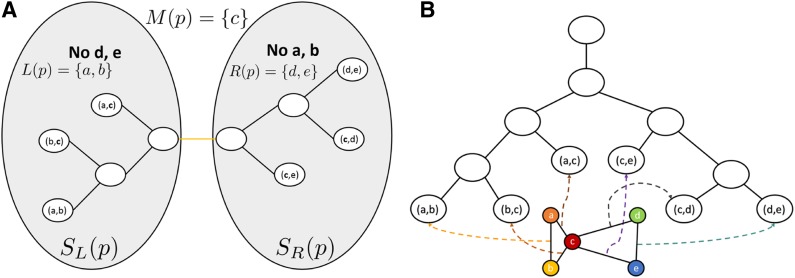
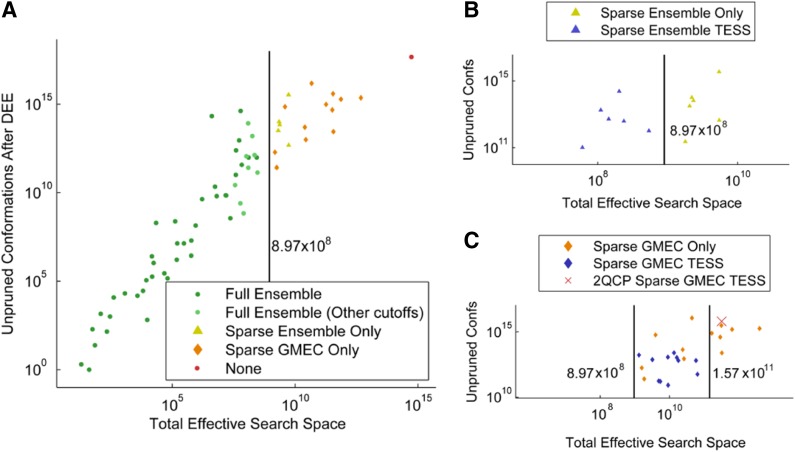
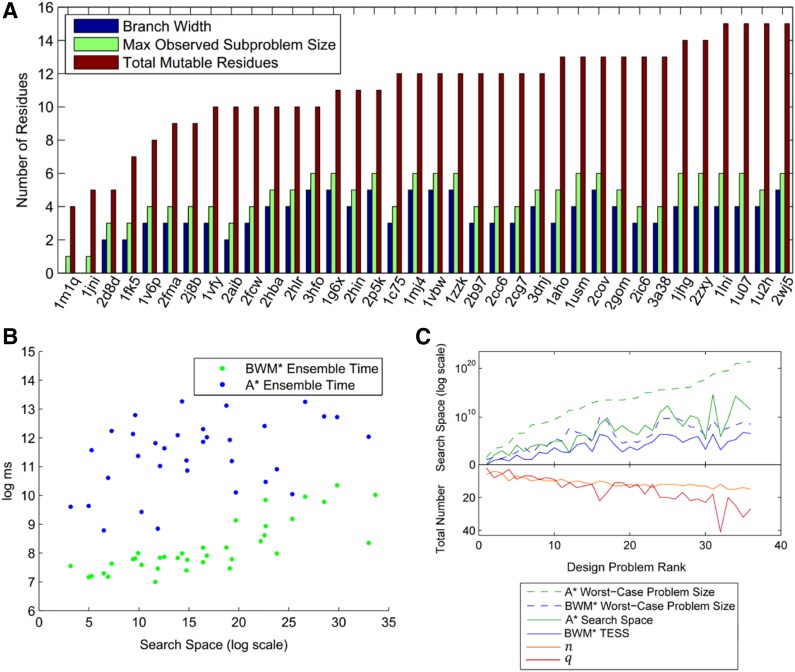
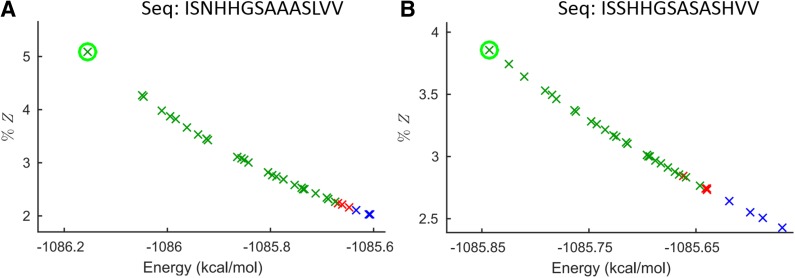
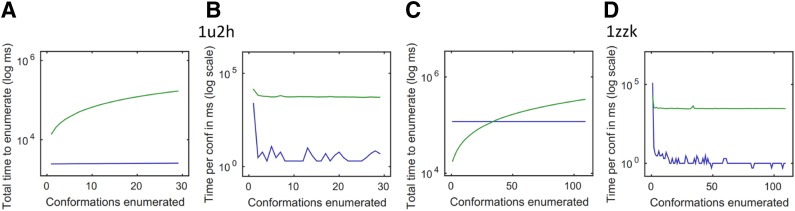
Sparse energy functions that ignore long range interactions between residue pairs are frequently used by protein design algorithms to reduce computational cost. Current dynamic programming algorithms that fully exploit the optimal substructure produced by these energy functions only compute the GMEC. This disproportionately favors the sequence of a single, static conformation and overlooks better binding sequences with multiple low-energy conformations. Provable, ensemble-based algorithms such as A* avoid this problem, but A* cannot guarantee better performance than exhaustive enumeration. We propose a novel, provable, dynamic programming algorithm called Branch-Width Minimization* (BWM*) to enumerate a gap-free ensemble of conformations in order of increasing energy. Given a branch-decomposition of branch-width w for an n-residue protein design with at most q discrete side-chain conformations per residue, BWM* returns the sparse GMEC in O([Formula: see text]) time and enumerates each additional conformation in merely O([Formula: see text]) time. We define a new measure, Total Effective Search Space (TESS), which can be computed efficiently a priori before BWM* or A* is run. We ran BWM* on 67 protein design problems and found that TESS discriminated between BWM*-efficient and A*-efficient cases with 100% accuracy. As predicted by TESS and validated experimentally, BWM* outperforms A* in 73% of the cases and computes the full ensemble or a close approximation faster than A*, enumerating each additional conformation in milliseconds. Unlike A*, the performance of BWM* can be predicted in polynomial time before running the algorithm, which gives protein designers the power to choose the most efficient algorithm for their particular design problem.
Keywords: OSPREY; branch-decomposition; dynamic programming; ensemble-based algorithms; protein design; provable algorithms; sparse residue interaction graphs.
Figures
References
-
- Dechter R., and Mateescu R. 2007. AND/OR search spaces for graphical models. Artif. Intell. 171, 73–106
-
- Desmet J., Spriet J., and Lasters I. 2002. Fast and accurate side-chain topology and energy refinement (FASTER) as a new method for protein structure optimization. Proteins 48, 31–43 - PubMed
-
- Donald B.R. 2011. Algorithms in Structural Molecular Biology. MIT Press, New York
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
