

Congenital insensitivity to pain: Fracturing without apparent skeletal pathobiology caused by an autosomal dominant, second mutation in SCN11A encoding voltage-gated sodium channel 1.9
- PMID: 26746779
- PMCID: PMC4755825
- DOI: 10.1016/j.bone.2015.11.022
Congenital insensitivity to pain: Fracturing without apparent skeletal pathobiology caused by an autosomal dominant, second mutation in SCN11A encoding voltage-gated sodium channel 1.9
Abstract
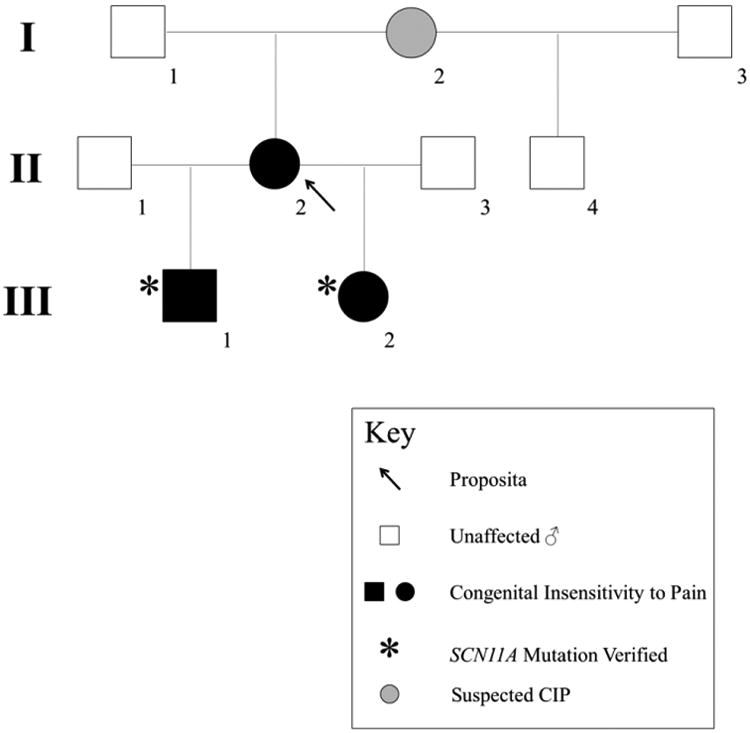
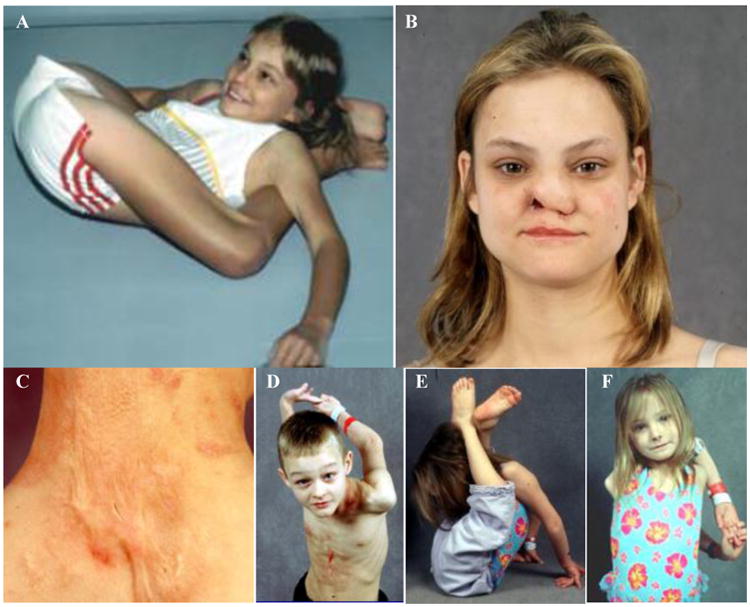
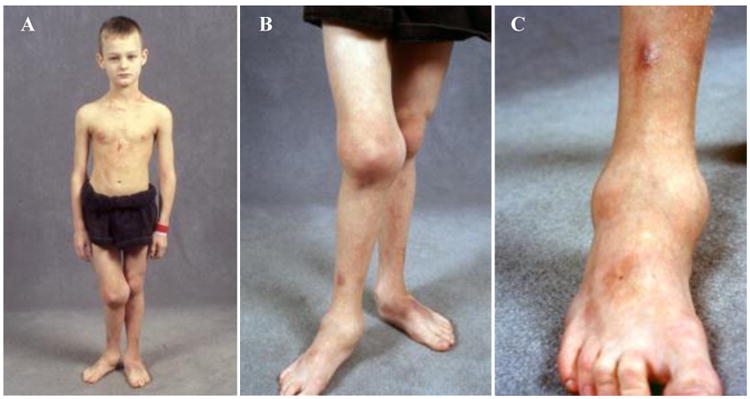
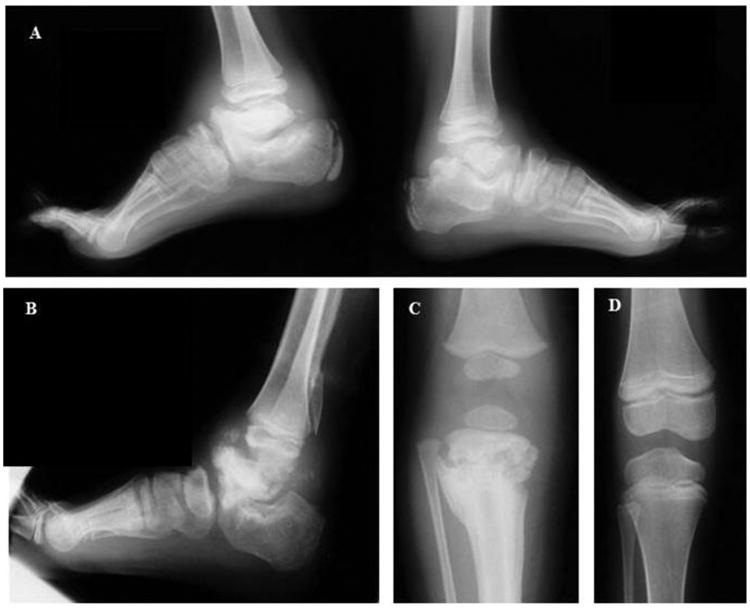
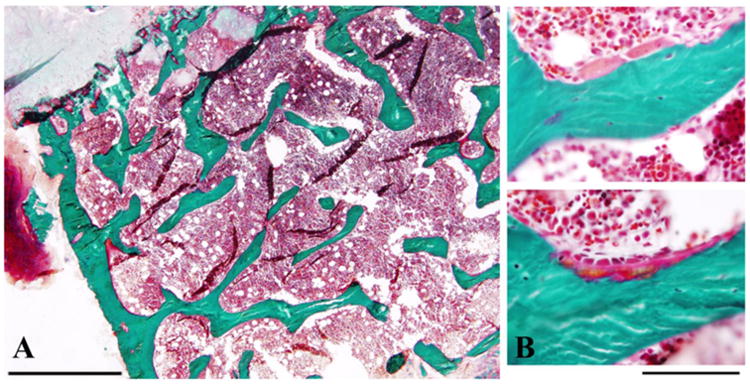
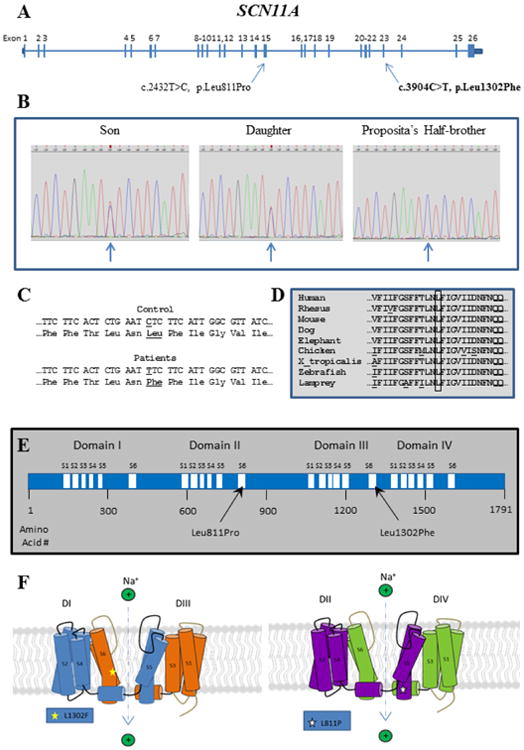
Congenital insensitivity to pain (CIP) comprises the rare heritable disorders without peripheral neuropathy that feature inability to feel pain. Fracturing and joint destruction are common complications, but lack detailed studies of mineral and skeletal homeostasis and bone histology. In 2013, discovery of a heterozygous gain-of-function mutation in SCN11A encoding voltage-gated sodium channel 1.9 (Nav1.9) established a distinctive CIP in three unrelated patients who suffered multiple painless fractures, self-inflicted mutilation, chronic diarrhea, and hyperhidrosis. Here, we studied a mother and two children with CIP by physical examination, biochemical testing, radiological imaging including DXA, iliac crest histology, and mutation analysis. She suffered fractures primarily of her lower extremities beginning at age two years, and had Charcot deformity of both ankles and joint hypermobility. Nerve conduction velocity together with electromyography were normal. Her children had recurrent major fractures beginning in early childhood, joint hypermobility, and chronic diarrhea. She had an excoriated external nare, and both children had hypertrophic scars from scratching. Skin collagen studies were normal. Radiographs revealed fractures and deformities. However, lumbar spine and total hip BMD Z-scores, biochemical parameters of mineral and skeletal homeostasis, and iliac crest histology of the mother (after in vivo tetracycline labeling) were normal. Genomic DNA from the children revealed a unique heterozygous missense mutation in exon 23 (c.3904C>T, p.Leu1302Phe) of SCN11A that is absent in SNP databases and alters an evolutionarily conserved amino acid. This autosomal dominant CIP reflects the second gain-of-function mutation of SCN11A. Perhaps joint hypermobility is an unreported feature. How mutation of Nav1.9 causes fracturing remains unexplained. Lack of injury awareness is typically offered as the reason, and was supported by our unremarkable biochemical, radiological, and histological findings indicating no skeletal pathobiology. However, low-trauma fracturing in these patients suggests an uncharacterized defect in bone quality.
Keywords: Charcot arthropathy; Joint hypermobility; Mineral homeostasis; Skeletal homeostasis.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
References
-
- Nagasako EM, Oaklander AL, Dworkin RH. Congenital insensitivity to pain: an update. Pain. 2003;101:213–9. - PubMed
-
- Dearborn G. A case of congenital general pure analgesia. J Nerv Ment Dis. 1932;75:612–5.
-
- Ford FR, Wilkins L. Congenital universal insensitiveness to pain. Bull Johns Hopkins Hosp. 1938;62:448–66.
-
- Boyd DA, Nie LW. Congenital universal indifference to pain. Arch Neurol Psychiatry. 1949;61:402–12. - PubMed
-
- Arbuse DI, Cantor MB, Barenberg PA. Congenital indifference to pain. J Pediatr. 1949;35(2):221–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
