

Loss of neutral ceramidase protects cells from nutrient- and energy -deprivation-induced cell death
- PMID: 26747710
- PMCID: PMC5513154
- DOI: 10.1042/BJ20150586
Loss of neutral ceramidase protects cells from nutrient- and energy -deprivation-induced cell death
Abstract
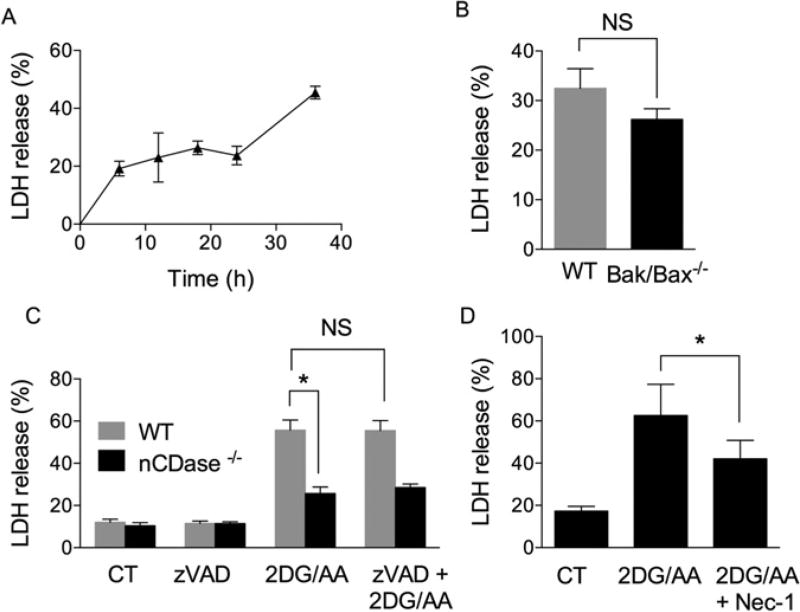
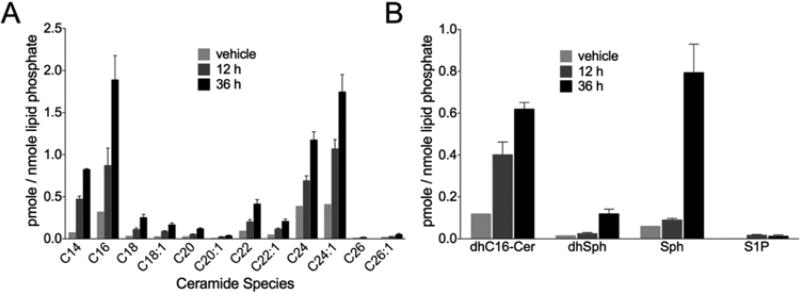
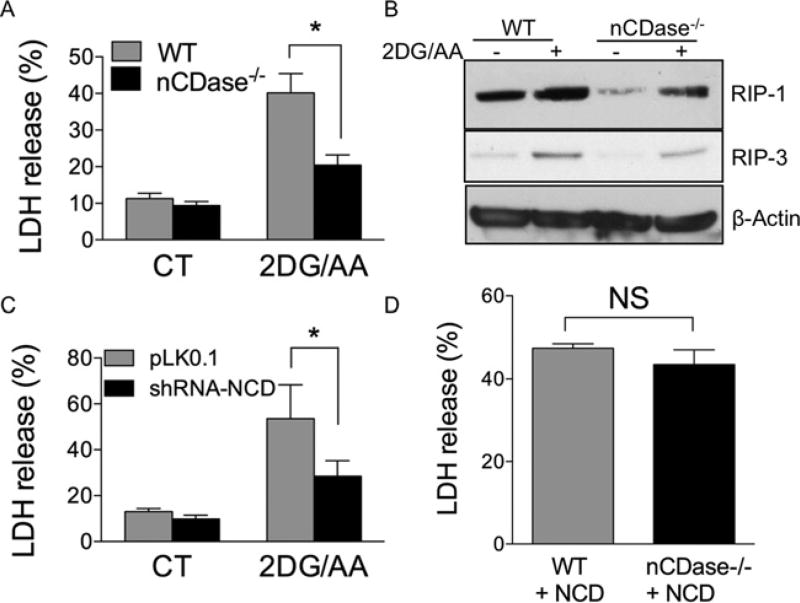
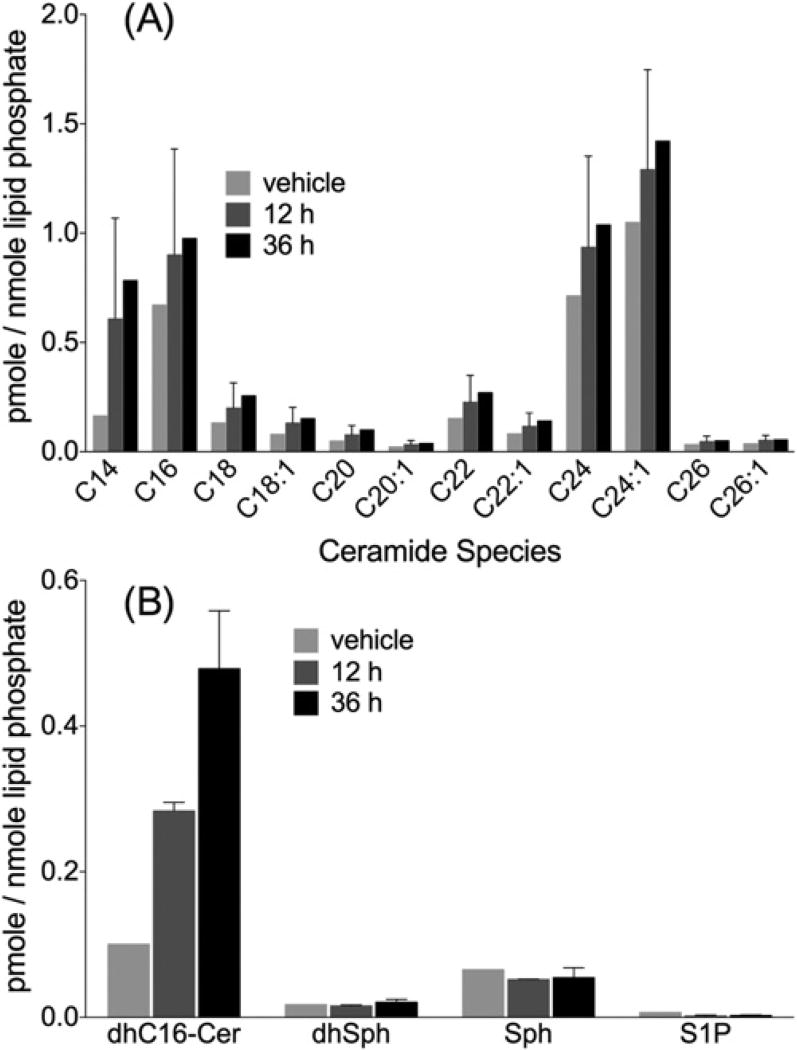
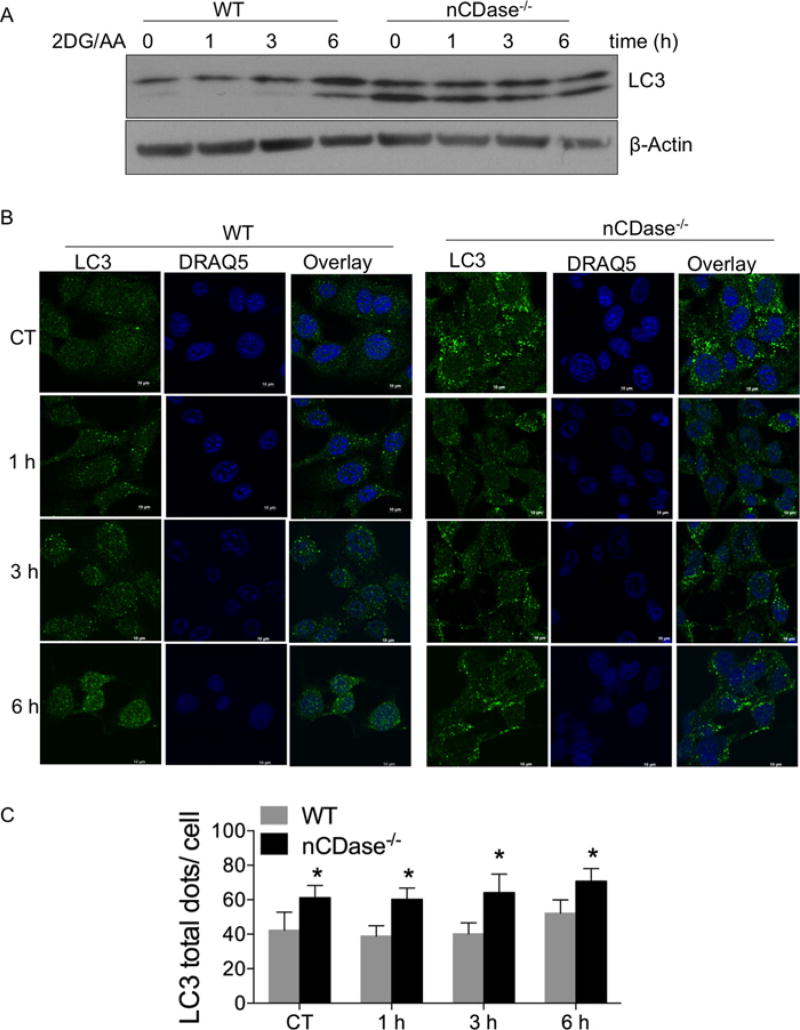
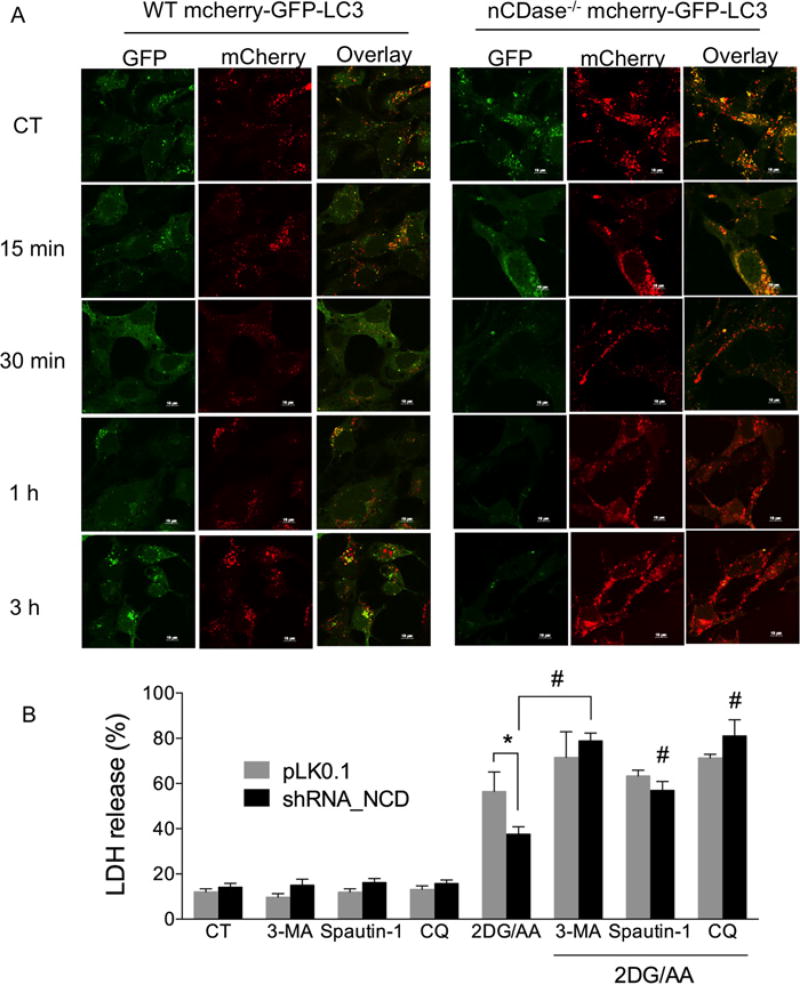
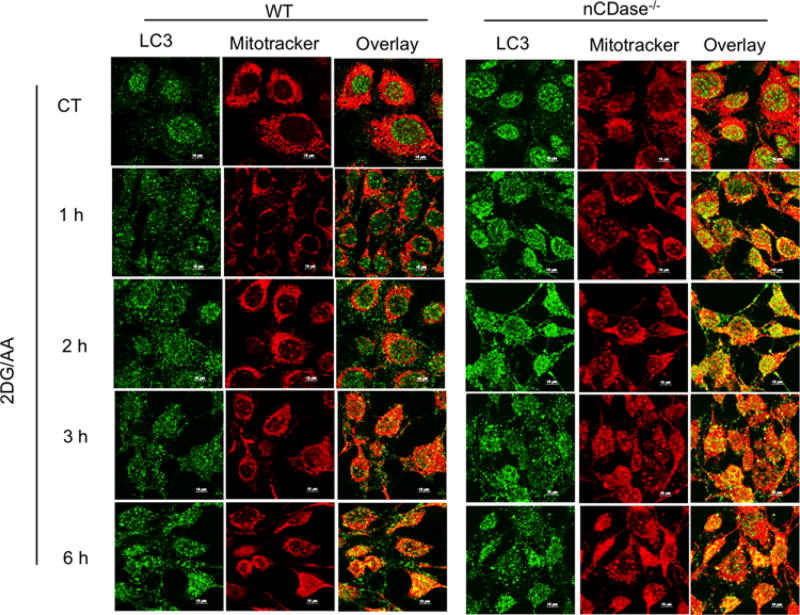
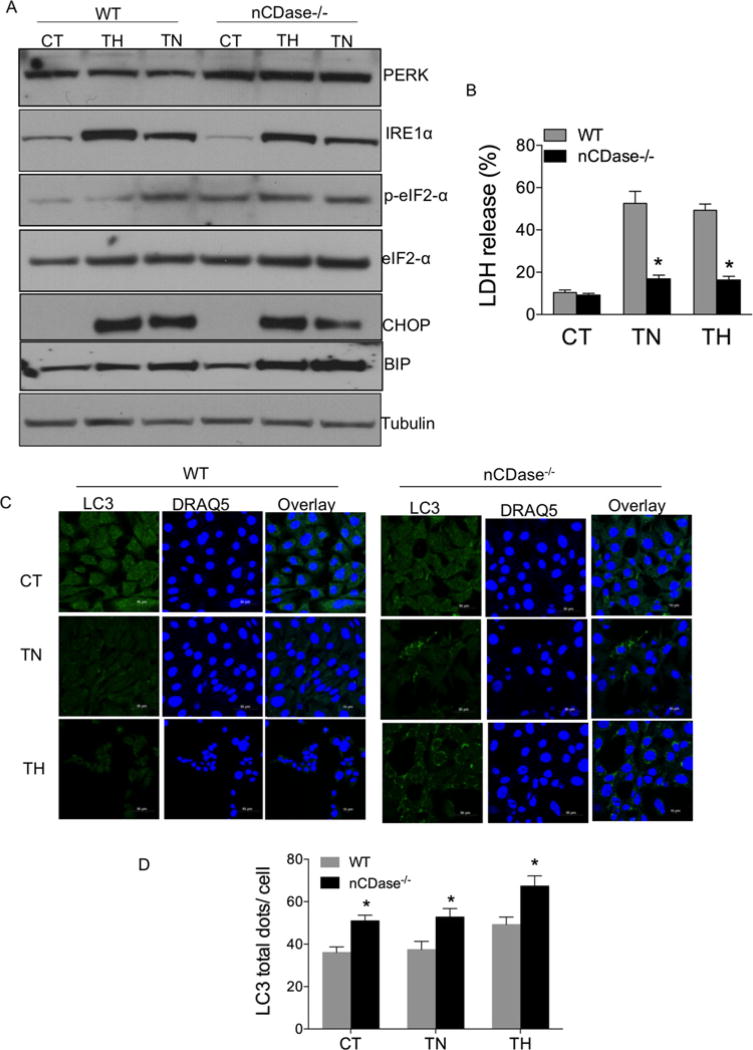
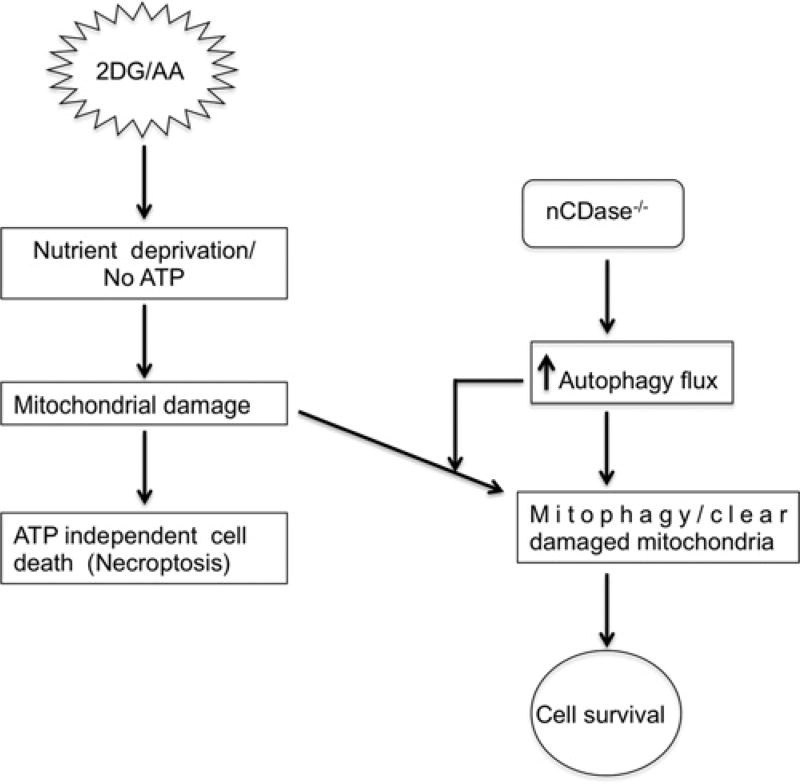
Sphingolipids are a family of lipids that regulate the cell cycle, differentiation and cell death. Sphingolipids are known to play a role in the induction of apoptosis, but a role for these lipids in necroptosis is largely unknown. Necroptosis is a programmed form of cell death that, unlike apoptosis, does not require ATP. Necroptosis can be induced under a variety of conditions, including nutrient deprivation and plays a major role in ischaemia/reperfusion injury to organs. Sphingolipids play a role in ischaemia/reperfusion injury in several organs. Thus, we hypothesized that sphingolipids mediate nutrient-deprivation-induced necroptosis. To address this, we utilized mouse embryonic fibroblast (MEFs) treated with 2-deoxyglucose (2DG) and antimycin A (AA) to inhibit glycolysis and mitochondrial electron transport. 2DG/AA treatment of MEFs induced necroptosis as it was receptor- interacting protein (RIP)-1/3 kinase-dependent and caspase-independent. Ceramides, sphingosine (Sph) and sphingosine 1-phosphate (S1P) were increased following 2DG/AA treatment. Cells lacking neutral ceramidase (nCDase(-/-)) were protected from 2DG/AA. Although nCDase(-/-) cells generated ceramides following 2DG/AA treatment, they did not generate Sph or S1P. This protection was stimulus-independent as nCDase(-/-) cells were also protected from endoplasmic reticulum (ER) stressors [tunicamycin (TN) or thapsigargin (TG)]. nCDase(-/-) MEFs had higher autophagic flux and mitophagy than wild-type (WT) MEFs and inhibition of autophagy sensitized them to necroptosis. These data indicate that loss of nCDase protects cells from nutrient- deprivation-induced necroptosis via autophagy, and clearance of damaged mitochondria. Results suggest that nCDase is a mediator of necroptosis and might be a novel therapeutic target for protection from ischaemic injury.
Keywords: autophagy flux; endoplasmic reticulum (ER) stress; mitophagy; necroptosis; neutral ceramidase; sphingolipids.
© 2016 Authors; published by Portland Press Limited.
Figures
References
-
- Orrenius S. Apoptosis: molecular mechanisms and implications for human disease. J. Intern. Med. 1995;237:529–536. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous