

Elucidating MicroRNA Regulatory Networks Using Transcriptional, Post-transcriptional, and Histone Modification Measurements
- PMID: 26748710
- PMCID: PMC4831719
- DOI: 10.1016/j.celrep.2015.12.031
Elucidating MicroRNA Regulatory Networks Using Transcriptional, Post-transcriptional, and Histone Modification Measurements
Abstract
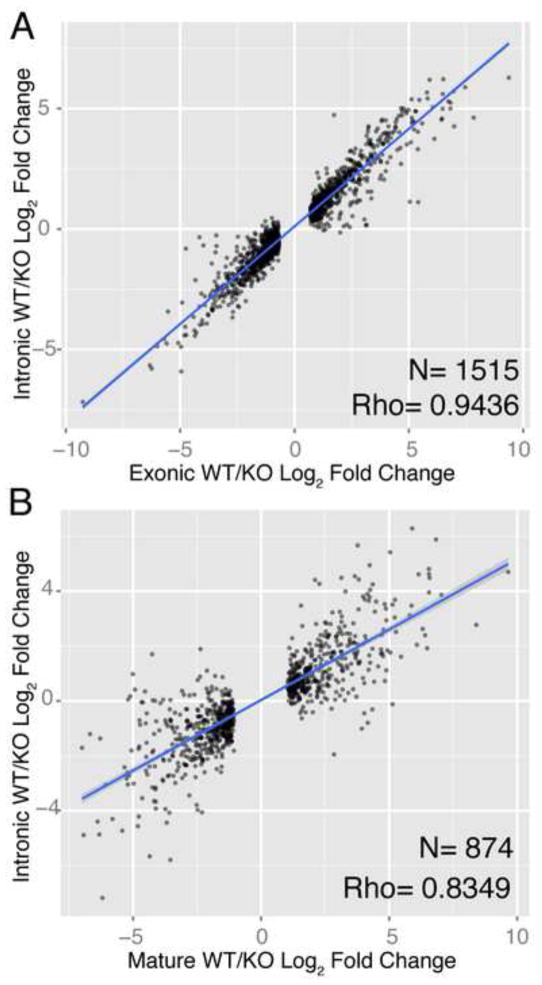
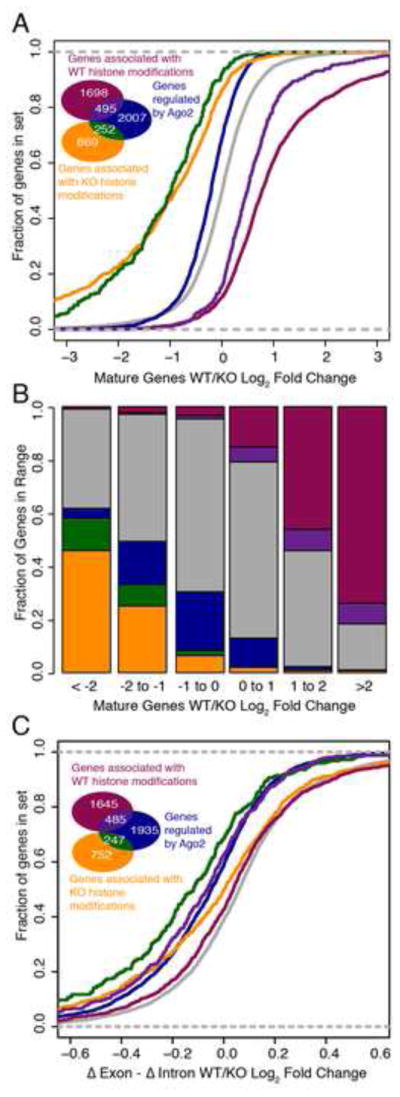
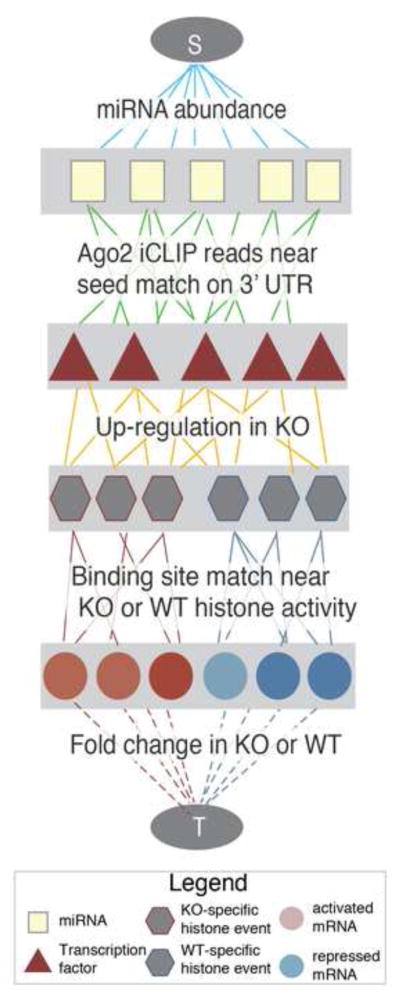
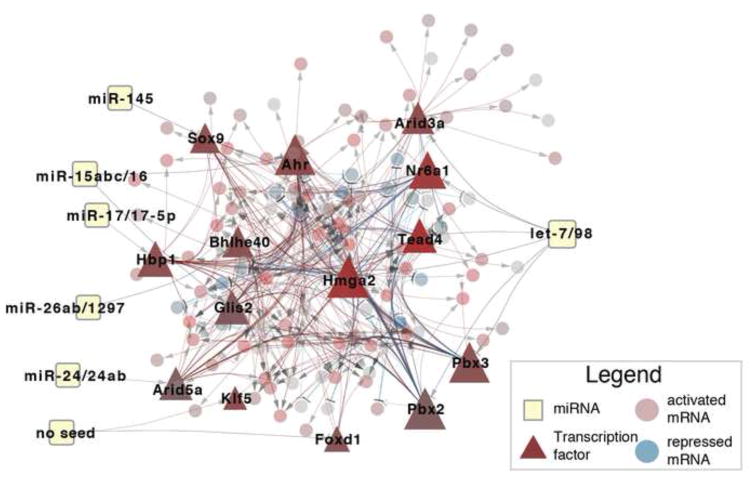
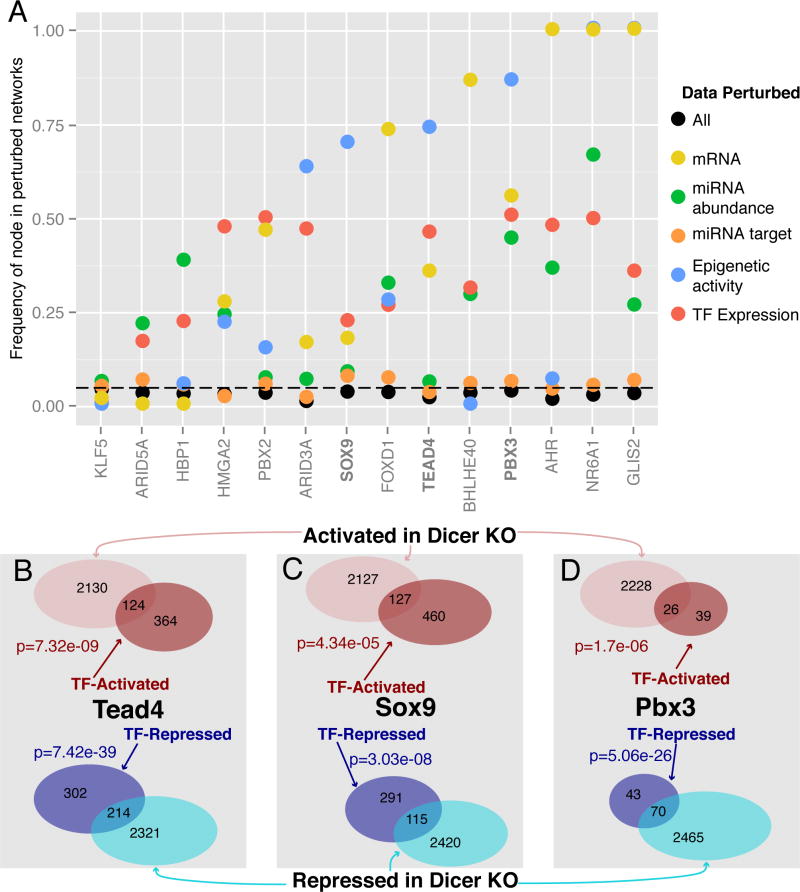
MicroRNAs (miRNAs) regulate diverse biological processes by repressing mRNAs, but their modest effects on direct targets, together with their participation in larger regulatory networks, make it challenging to delineate miRNA-mediated effects. Here, we describe an approach to characterizing miRNA-regulatory networks by systematically profiling transcriptional, post-transcriptional and epigenetic activity in a pair of isogenic murine fibroblast cell lines with and without Dicer expression. By RNA sequencing (RNA-seq) and CLIP (crosslinking followed by immunoprecipitation) sequencing (CLIP-seq), we found that most of the changes induced by global miRNA loss occur at the level of transcription. We then introduced a network modeling approach that integrated these data with epigenetic data to identify specific miRNA-regulated transcription factors that explain the impact of miRNA perturbation on gene expression. In total, we demonstrate that combining multiple genome-wide datasets spanning diverse regulatory modes enables accurate delineation of the downstream miRNA-regulated transcriptional network and establishes a model for studying similar networks in other systems.
Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, Farnham PJ, Hirst M, Lander ES, Mikkelsen TS, Thomson JA. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28:1045–8. doi: 10.1038/nbt1010-1045. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
