

Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA
- PMID: 26749308
- PMCID: PMC4716689
- DOI: 10.1016/j.ajhg.2015.11.023
Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA
Abstract
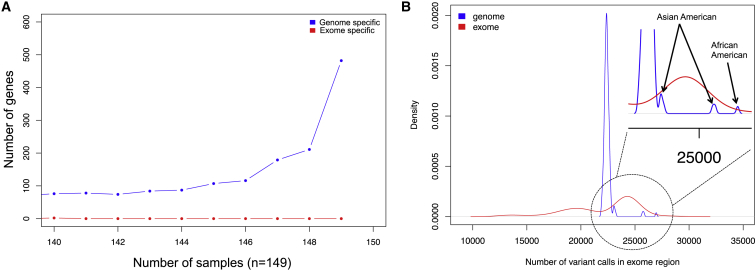
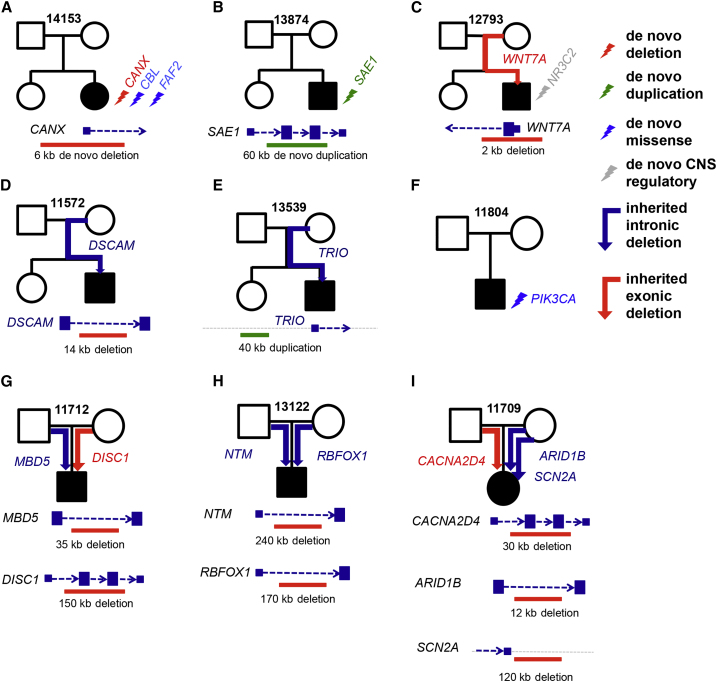
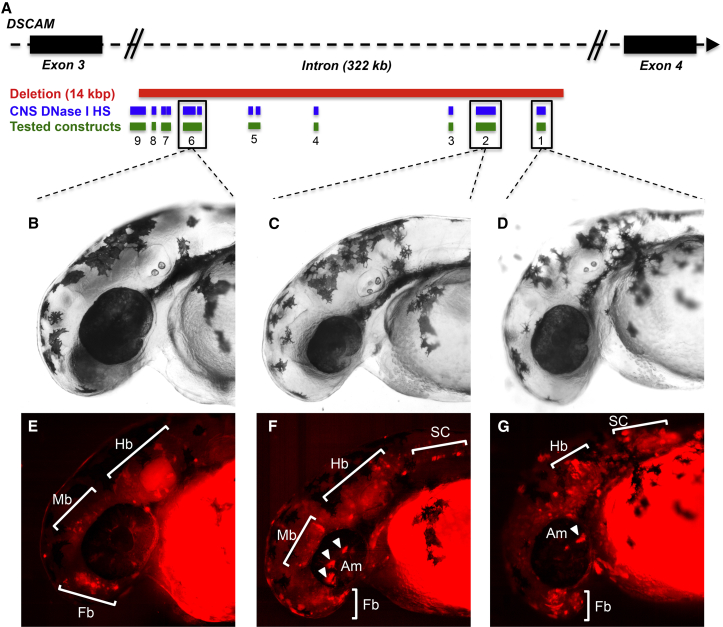
We performed whole-genome sequencing (WGS) of 208 genomes from 53 families affected by simplex autism. For the majority of these families, no copy-number variant (CNV) or candidate de novo gene-disruptive single-nucleotide variant (SNV) had been detected by microarray or whole-exome sequencing (WES). We integrated multiple CNV and SNV analyses and extensive experimental validation to identify additional candidate mutations in eight families. We report that compared to control individuals, probands showed a significant (p = 0.03) enrichment of de novo and private disruptive mutations within fetal CNS DNase I hypersensitive sites (i.e., putative regulatory regions). This effect was only observed within 50 kb of genes that have been previously associated with autism risk, including genes where dosage sensitivity has already been established by recurrent disruptive de novo protein-coding mutations (ARID1B, SCN2A, NR3C2, PRKCA, and DSCAM). In addition, we provide evidence of gene-disruptive CNVs (in DISC1, WNT7A, RBFOX1, and MBD5), as well as smaller de novo CNVs and exon-specific SNVs missed by exome sequencing in neurodevelopmental genes (e.g., CANX, SAE1, and PIK3CA). Our results suggest that the detection of smaller, often multiple CNVs affecting putative regulatory elements might help explain additional risk of simplex autism.
Copyright © 2016 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Steffenburg S., Gillberg C., Hellgren L., Andersson L., Gillberg I.C., Jakobsson G., Bohman M. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J. Child Psychol. Psychiatry. 1989;30:405–416. - PubMed
-
- Bailey A., Le Couteur A., Gottesman I., Bolton P., Simonoff E., Yuzda E., Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol. Med. 1995;25:63–77. - PubMed
-
- Verkerk A.J., Pieretti M., Sutcliffe J.S., Fu Y.H., Kuhl D.P., Pizzuti A., Reiner O., Richards S., Victoria M.F., Zhang F.P. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. - PubMed
-
- Amir R.E., Van den Veyver I.B., Wan M., Tran C.Q., Francke U., Zoghbi H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–188. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U54 HD083091/HD/NICHD NIH HHS/United States
- HHSN267200700021C/HD/NICHD NIH HHS/United States
- T32 GM007814/GM/NIGMS NIH HHS/United States
- HHSN275200800015C/HD/NICHD NIH HHS/United States
- 5T32GM07814/GM/NIGMS NIH HHS/United States
- UM1 HG008901/HG/NHGRI NIH HHS/United States
- T32 HG000035/HG/NHGRI NIH HHS/United States
- 2T32HG000035/HG/NHGRI NIH HHS/United States
- R01 NS062972/NS/NINDS NIH HHS/United States
- NS062972/NS/NINDS NIH HHS/United States
- HHSN267200700023C/HD/NICHD NIH HHS/United States
- Howard Hughes Medical Institute/United States
- T32 GM007266/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
