

Autosomal-Dominant Corneal Endothelial Dystrophies CHED1 and PPCD1 Are Allelic Disorders Caused by Non-coding Mutations in the Promoter of OVOL2
- PMID: 26749309
- PMCID: PMC4716680
- DOI: 10.1016/j.ajhg.2015.11.018
Autosomal-Dominant Corneal Endothelial Dystrophies CHED1 and PPCD1 Are Allelic Disorders Caused by Non-coding Mutations in the Promoter of OVOL2
Abstract
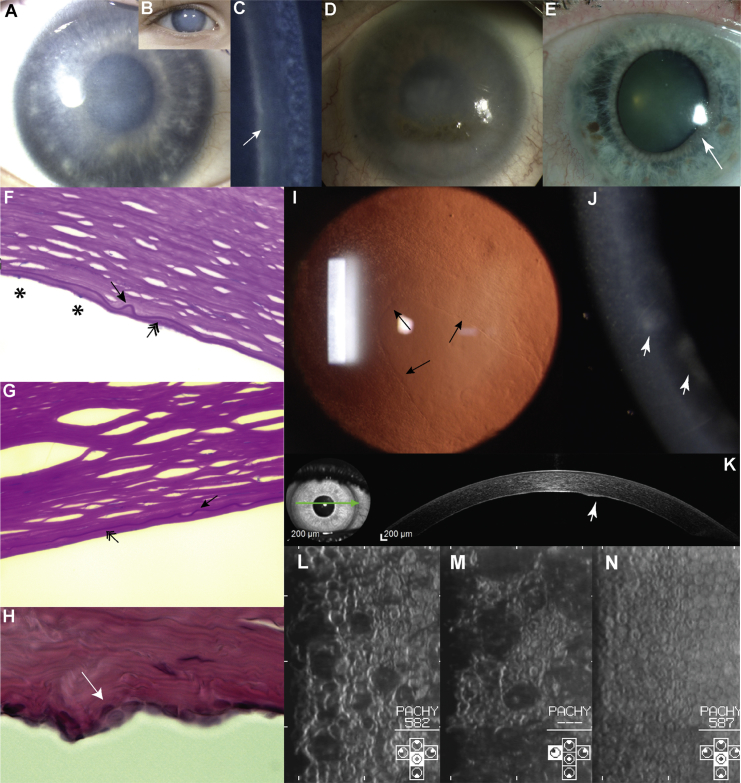
Congenital hereditary endothelial dystrophy 1 (CHED1) and posterior polymorphous corneal dystrophy 1 (PPCD1) are autosomal-dominant corneal endothelial dystrophies that have been genetically mapped to overlapping loci on the short arm of chromosome 20. We combined genetic and genomic approaches to identify the cause of disease in extensive pedigrees comprising over 100 affected individuals. After exclusion of pathogenic coding, splice-site, and copy-number variations, a parallel approach using targeted and whole-genome sequencing facilitated the identification of pathogenic variants in a conserved region of the OVOL2 proximal promoter sequence in the index families (c.-339_361dup for CHED1 and c.-370T>C for PPCD1). Direct sequencing of the OVOL2 promoter in other unrelated affected individuals identified two additional mutations within the conserved proximal promoter sequence (c.-274T>G and c.-307T>C). OVOL2 encodes ovo-like zinc finger 2, a C2H2 zinc-finger transcription factor that regulates mesenchymal-to-epithelial transition and acts as a direct transcriptional repressor of the established PPCD-associated gene ZEB1. Interestingly, we did not detect OVOL2 expression in the normal corneal endothelium. Our in vitro data demonstrate that all four mutated OVOL2 promoters exhibited more transcriptional activity than the corresponding wild-type promoter, and we postulate that the mutations identified create cryptic cis-acting regulatory sequence binding sites that drive aberrant OVOL2 expression during endothelial cell development. Our data establish CHED1 and PPCD1 as allelic conditions and show that CHED1 represents the extreme of what can be considered a disease spectrum. They also implicate transcriptional dysregulation of OVOL2 as a common cause of dominantly inherited corneal endothelial dystrophies.
Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Figures



References
-
- Weiss J.S., Møller H.U., Aldave A.J., Seitz B., Bredrup C., Kivelä T., Munier F.L., Rapuano C.J., Nischal K.K., Kim E.K. IC3D classification of corneal dystrophies--edition 2. Cornea. 2015;34:117–159. - PubMed
-
- Cibis G.W., Krachmer J.A., Phelps C.D., Weingeist T.A. The clinical spectrum of posterior polymorphous dystrophy. Arch. Ophthalmol. 1977;95:1529–1537. - PubMed
-
- Yellore V.S., Papp J.C., Sobel E., Khan M.A., Rayner S.A., Farber D.B., Aldave A.J. Replication and refinement of linkage of posterior polymorphous corneal dystrophy to the posterior polymorphous corneal dystrophy 1 locus on chromosome 20. Genet. Med. 2007;9:228–234. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
