

Exploring the genetic basis of early-onset chronic kidney disease
- PMID: 26750453
- PMCID: PMC5202482
- DOI: 10.1038/nrneph.2015.205
Exploring the genetic basis of early-onset chronic kidney disease
Abstract
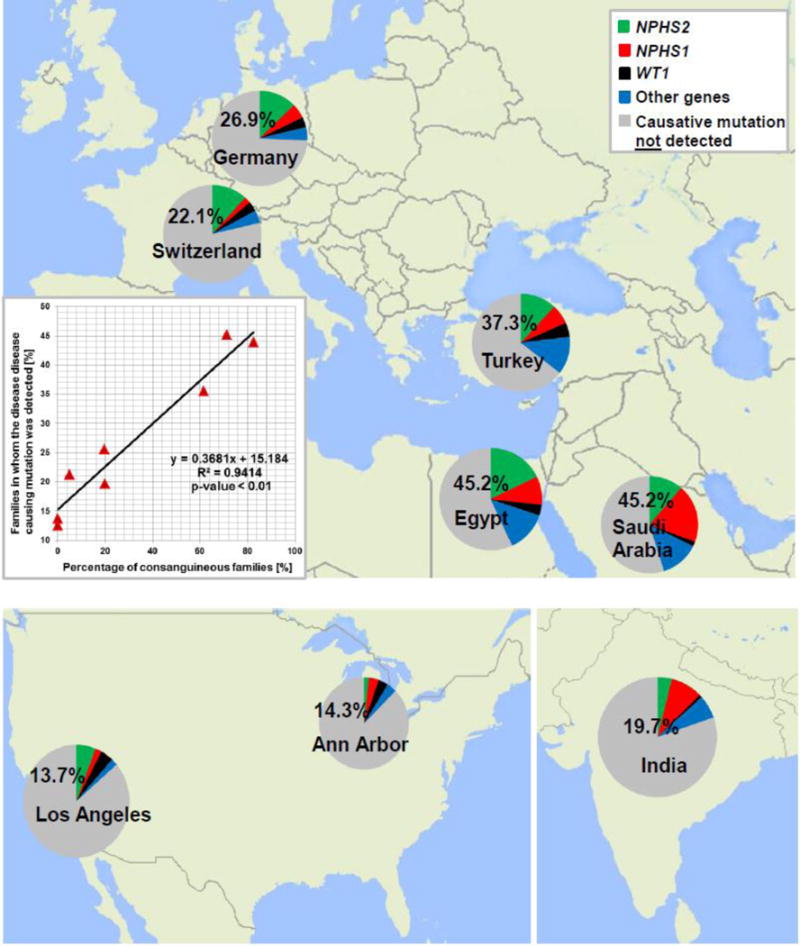
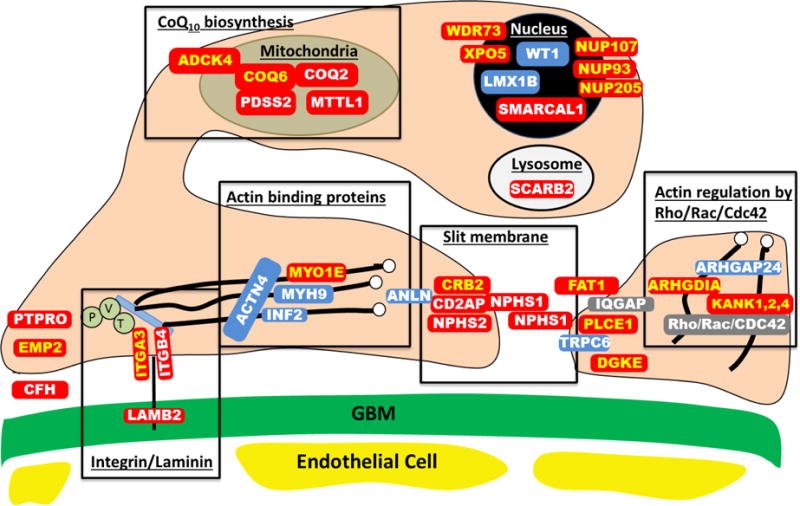
The primary causes of chronic kidney disease (CKD) in children differ from those of CKD in adults. In the USA the most common diagnostic groups of renal disease that manifest before the age of 25 years are congenital anomalies of the kidneys and urinary tract, steroid-resistant nephrotic syndrome, chronic glomerulonephritis and renal cystic ciliopathies, which together encompass >70% of early-onset CKD diagnoses. Findings from the past decade suggest that early-onset CKD is caused by mutations in any one of over 200 different monogenic genes. Developments in high-throughput sequencing in the past few years has rendered identification of causative mutations in this high number of genes feasible. Use of genetic analyses in patients with early onset-CKD will provide patients and their families with a molecular genetic diagnosis, generate new insights into disease mechanisms, facilitate aetiology-based classifications of patient cohorts for clinical studies, and might have consequences for personalized approaches to the prevention and treatment of CKD. In this Review, we discuss the implications of next-generation sequencing in clinical genetic diagnostics and the discovery of novel genes in early-onset CKD. We also delineate the resulting opportunities for deciphering disease mechanisms and the therapeutic implications of these findings.
Figures
References
-
- Inker LA, et al. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for the evaluation and management of CKD. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2014;63:713–735. - PubMed
-
- Coresh J, et al. Prevalence of chronic kidney disease in the United States. Jama. 2007;298:2038–2047. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
