

Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice
- PMID: 26752519
- PMCID: PMC4742374
- DOI: 10.1038/nm.4021
Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice
Abstract
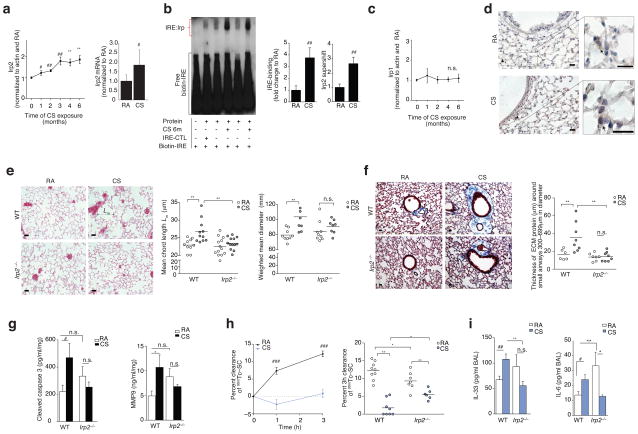
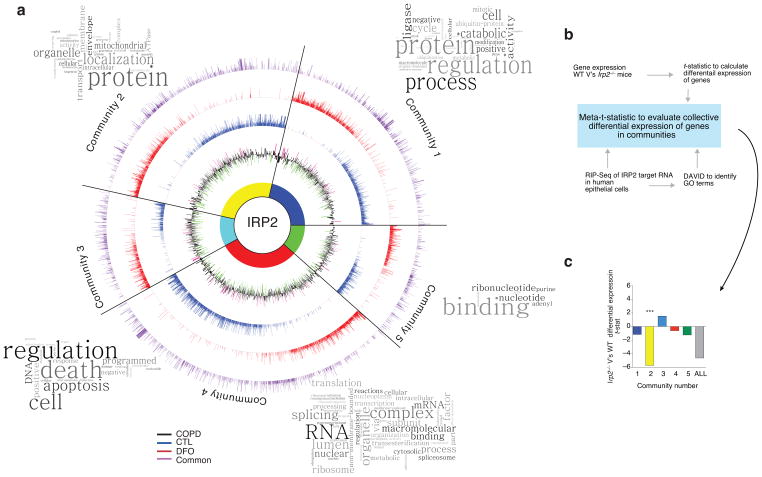
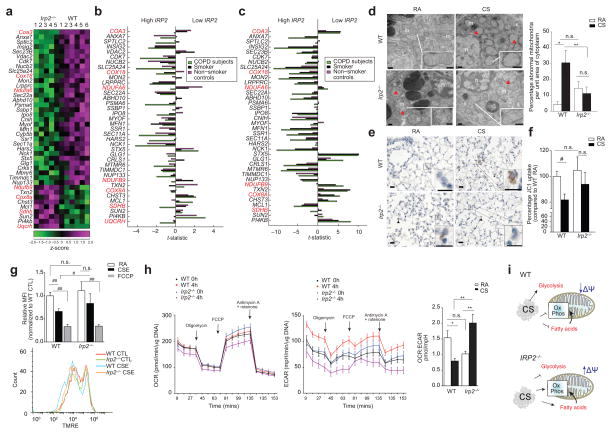
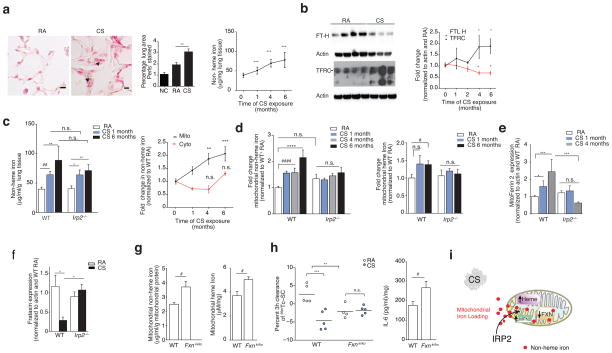
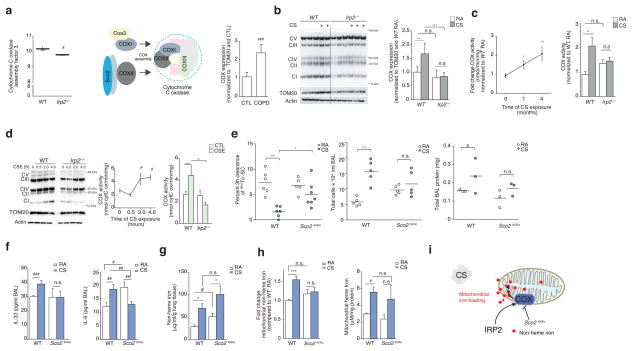
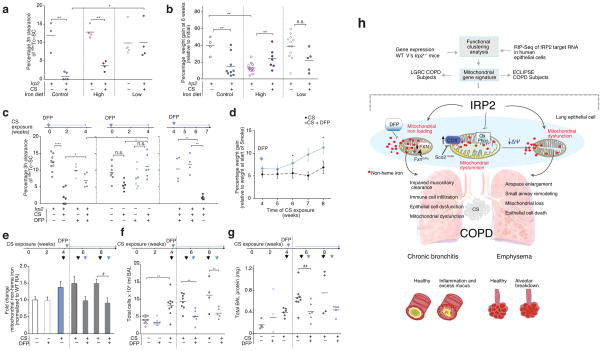
Chronic obstructive pulmonary disease (COPD) is linked to both cigarette smoking and genetic determinants. We have previously identified iron-responsive element-binding protein 2 (IRP2) as an important COPD susceptibility gene and have shown that IRP2 protein is increased in the lungs of individuals with COPD. Here we demonstrate that mice deficient in Irp2 were protected from cigarette smoke (CS)-induced experimental COPD. By integrating RNA immunoprecipitation followed by sequencing (RIP-seq), RNA sequencing (RNA-seq), and gene expression and functional enrichment clustering analysis, we identified Irp2 as a regulator of mitochondrial function in the lungs of mice. Irp2 increased mitochondrial iron loading and levels of cytochrome c oxidase (COX), which led to mitochondrial dysfunction and subsequent experimental COPD. Frataxin-deficient mice, which had higher mitochondrial iron loading, showed impaired airway mucociliary clearance (MCC) and higher pulmonary inflammation at baseline, whereas mice deficient in the synthesis of cytochrome c oxidase, which have reduced COX, were protected from CS-induced pulmonary inflammation and impairment of MCC. Mice treated with a mitochondrial iron chelator or mice fed a low-iron diet were protected from CS-induced COPD. Mitochondrial iron chelation also alleviated CS-induced impairment of MCC, CS-induced pulmonary inflammation and CS-associated lung injury in mice with established COPD, suggesting a critical functional role and potential therapeutic intervention for the mitochondrial-iron axis in COPD.
Conflict of interest statement
In the past three years, Edwin K. Silverman received honoraria and consulting fees from Merck and grant support and consulting fees from GlaxoSmithKline.
Figures
Comment in
-
Respiratory disorders: Ironing out smoking-related airway disease.Nature. 2016 Mar 31;531(7596):586-7. doi: 10.1038/nature17309. Epub 2016 Mar 9. Nature. 2016. PMID: 26958829 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- R01-HL111759/HL/NHLBI NIH HHS/United States
- R21 ES025379/ES/NIEHS NIH HHS/United States
- R01-HL079904/HL/NHLBI NIH HHS/United States
- P01-HD080642/HD/NICHD NIH HHS/United States
- P01-HL114501/HL/NHLBI NIH HHS/United States
- HL122513/HL/NHLBI NIH HHS/United States
- K99 HL125899/HL/NHLBI NIH HHS/United States
- R01 HL132198/HL/NHLBI NIH HHS/United States
- R01-GM088999/GM/NIGMS NIH HHS/United States
- R01 AI111475/AI/NIAID NIH HHS/United States
- R01 HL079904/HL/NHLBI NIH HHS/United States
- R01 HL086814/HL/NHLBI NIH HHS/United States
- P01-HL105339/HL/NHLBI NIH HHS/United States
- R00 HL125899/HL/NHLBI NIH HHS/United States
- K99 HL122513/HL/NHLBI NIH HHS/United States
- K99-HL125899/HL/NHLBI NIH HHS/United States
- R21-HL111835/HL/NHLBI NIH HHS/United States
- R01-HL055330/HL/NHLBI NIH HHS/United States
- R00 HL122513/HL/NHLBI NIH HHS/United States
- R01 HL086936/HL/NHLBI NIH HHS/United States
- ImNIH/Intramural NIH HHS/United States
- P01 HD080642/HD/NICHD NIH HHS/United States
- R21-ES025379-01/ES/NIEHS NIH HHS/United States
- R01-AI111475-01/AI/NIAID NIH HHS/United States
- P01 HL105339/HL/NHLBI NIH HHS/United States
- R01 GM088999/GM/NIGMS NIH HHS/United States
- R01 HL055330/HL/NHLBI NIH HHS/United States
- R21 HL111835/HL/NHLBI NIH HHS/United States
- R01-HL086936/HL/NHLBI NIH HHS/United States
- P01 HD032062/HD/NICHD NIH HHS/United States
- P01 HL114501/HL/NHLBI NIH HHS/United States
- R01-HL86814/HL/NHLBI NIH HHS/United States
- R01 HL111759/HL/NHLBI NIH HHS/United States
- P30 ES009089/ES/NIEHS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
