

The importance of genetic diagnosis for Duchenne muscular dystrophy
- PMID: 26754139
- PMCID: PMC4789806
- DOI: 10.1136/jmedgenet-2015-103387
The importance of genetic diagnosis for Duchenne muscular dystrophy
Abstract
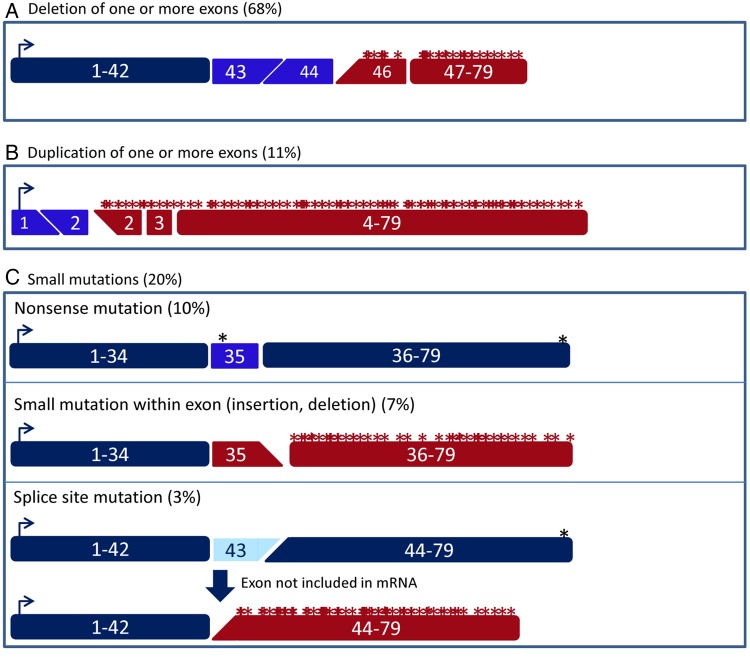
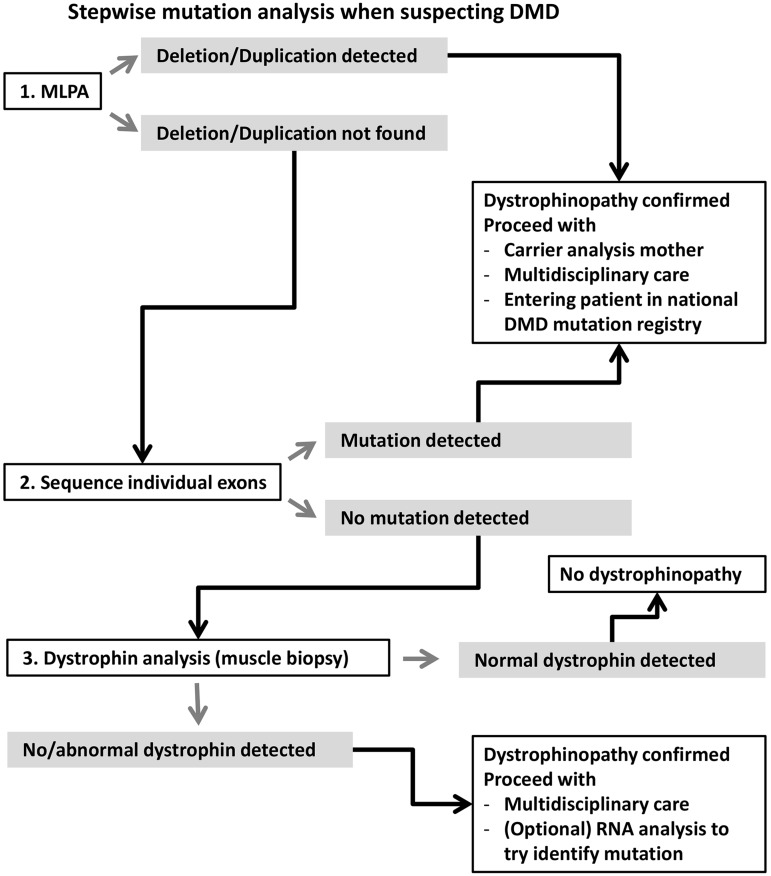
Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy are caused by mutations in the dystrophin-encoding DMD gene. Large deletions and duplications are most common, but small mutations have been found as well. Having a correct diagnosis is important for family planning and providing proper care to patients according to published guidelines. With mutation-specific therapies under development for DMD, a correct diagnosis is now also important for assessing whether patients are eligible for treatments. This review discusses different mutations causing DMD, diagnostic techniques available for making a genetic diagnosis for children suspected of DMD and the importance of having a specific genetic diagnosis in the context of emerging genetic therapies for DMD.
Keywords: Diagnosis; Genetics; Muscle disease.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://www.bmj.com/company/products-services/rights-and-licensing/
Figures

References
-
- Aartsma-Rus A. Dystrophin analysis in clinical trials. J Neuromuscul Dis 2014;1:41–53. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources