

Spinal cord injury-induced immune deficiency syndrome enhances infection susceptibility dependent on lesion level
- PMID: 26754788
- PMCID: PMC5014125
- DOI: 10.1093/brain/awv375
Spinal cord injury-induced immune deficiency syndrome enhances infection susceptibility dependent on lesion level
Abstract
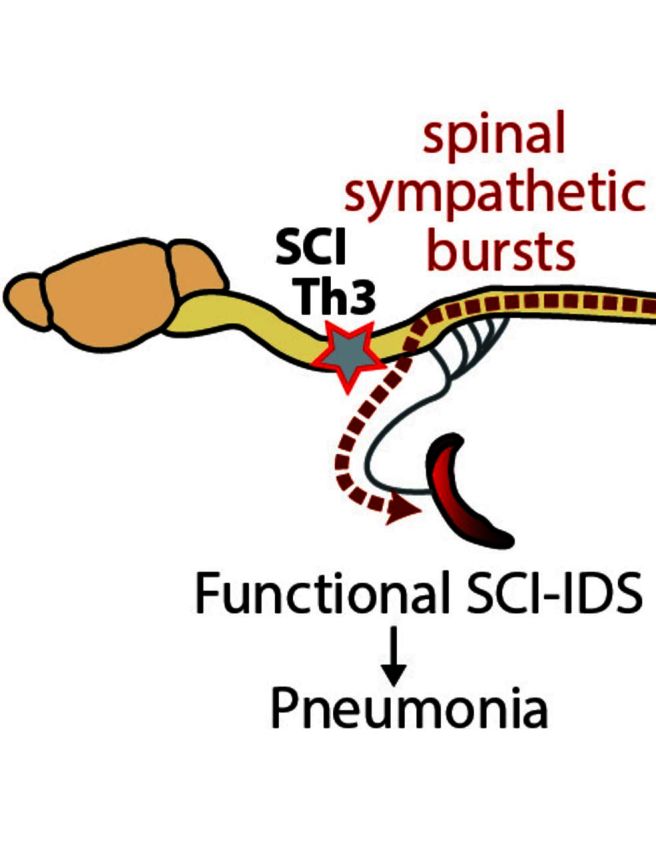
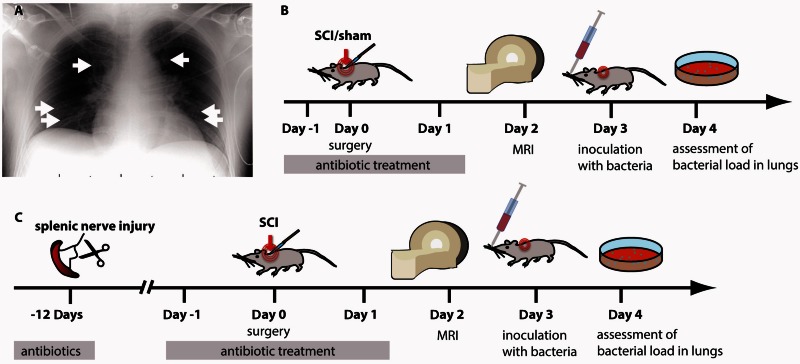
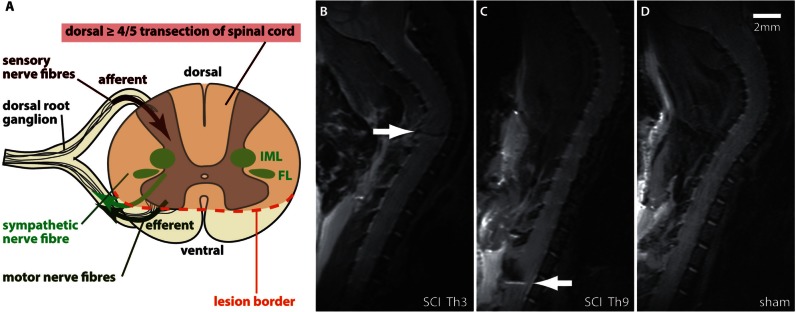
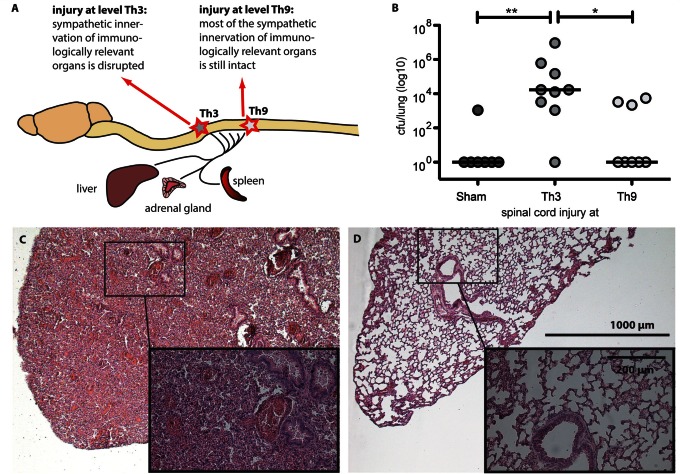
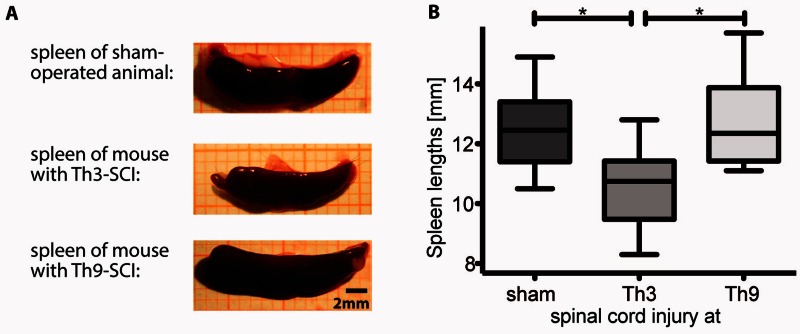
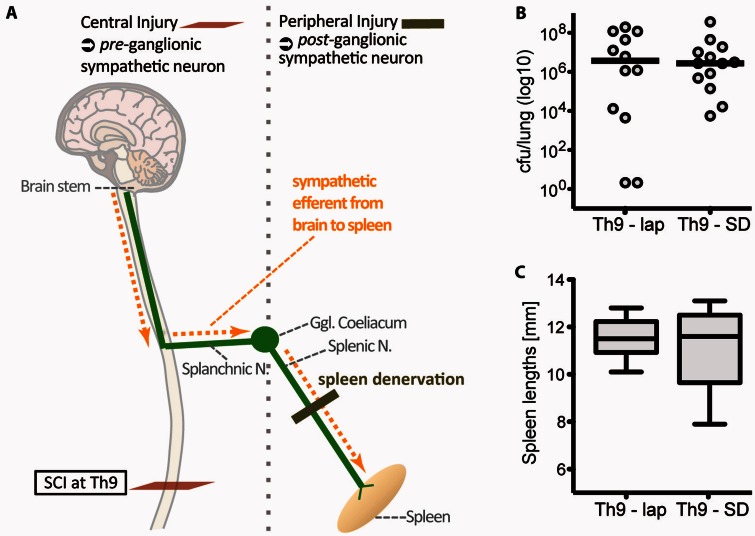
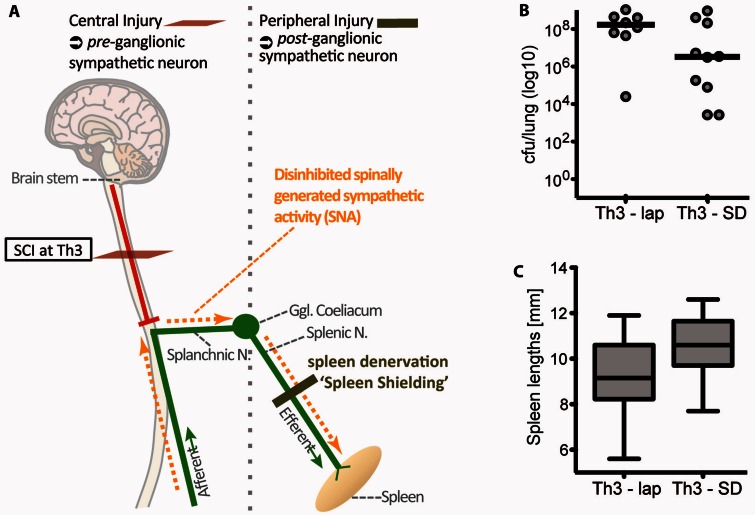
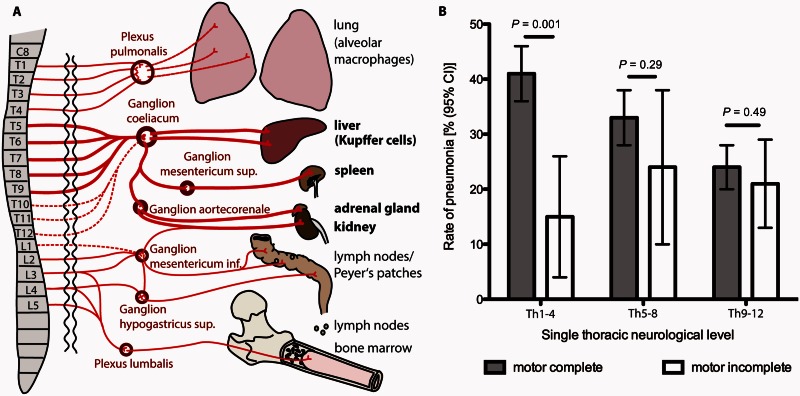
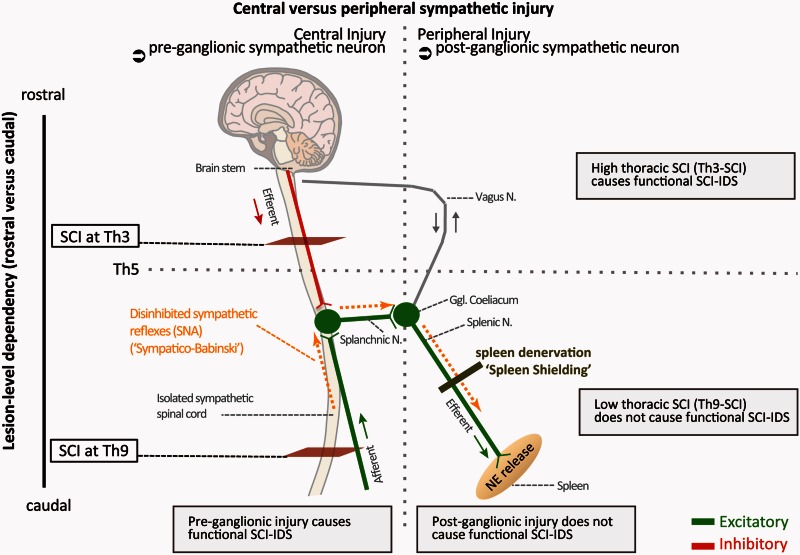
Pneumonia is the leading cause of death after acute spinal cord injury and is associated with poor neurological outcome. In contrast to the current understanding, attributing enhanced infection susceptibility solely to the patient's environment and motor dysfunction, we investigate whether a secondary functional neurogenic immune deficiency (spinal cord injury-induced immune deficiency syndrome, SCI-IDS) may account for the enhanced infection susceptibility. We applied a clinically relevant model of experimental induced pneumonia to investigate whether the systemic SCI-IDS is functional sufficient to cause pneumonia dependent on spinal cord injury lesion level and investigated whether findings are mirrored in a large prospective cohort study after human spinal cord injury. In a mouse model of inducible pneumonia, high thoracic lesions that interrupt sympathetic innervation to major immune organs, but not low thoracic lesions, significantly increased bacterial load in lungs. The ability to clear the bacterial load from the lung remained preserved in sham animals. Propagated immune susceptibility depended on injury of central pre-ganglionic but not peripheral postganglionic sympathetic innervation to the spleen. Thoracic spinal cord injury level was confirmed as an independent increased risk factor of pneumonia in patients after motor complete spinal cord injury (odds ratio = 1.35, P < 0.001) independently from mechanical ventilation and preserved sensory function by multiple regression analysis. We present evidence that spinal cord injury directly causes increased risk for bacterial infection in mice as well as in patients. Besides obvious motor and sensory paralysis, spinal cord injury also induces a functional SCI-IDS ('immune paralysis'), sufficient to propagate clinically relevant infection in an injury level dependent manner.
Keywords: mechanisms; myelopathy; neuroinflammation; rehabilitation; spinal cord injury.
© The Author (2016). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Ankeny DP, Lucin KM, Sanders VM, McGaughy VM, Popovich PG. Spinal cord injury triggers systemic autoimmunity: evidence for chronic B lymphocyte activation and lupus-like autoantibody synthesis. J Neurochem 2006; 99: 1073–87. - PubMed
-
- Campagnolo DI, Bartlett JA, Keller SE, Sanchez W, Oza R. Impaired phagocytosis of Staphylococcus aureus in complete tetraplegics. Am J Phys Med Rehabil 1997; 76: 276–80. - PubMed
-
- Candiani G, Abbondi M, Borgonovi M, Williams R. Experimental lobar pneumonia due to penicillin-susceptible and penicillin-resistant Streptococcus pneumoniae in immunocompetent and neutropenic rats: efficacy of penicillin and teicoplanin treatment. J Antimicrob Chemother 1997; 39: 199–207. - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
