

Analysis of TcdB Proteins within the Hypervirulent Clade 2 Reveals an Impact of RhoA Glucosylation on Clostridium difficile Proinflammatory Activities
- PMID: 26755157
- PMCID: PMC4771349
- DOI: 10.1128/IAI.01291-15
Analysis of TcdB Proteins within the Hypervirulent Clade 2 Reveals an Impact of RhoA Glucosylation on Clostridium difficile Proinflammatory Activities
Abstract
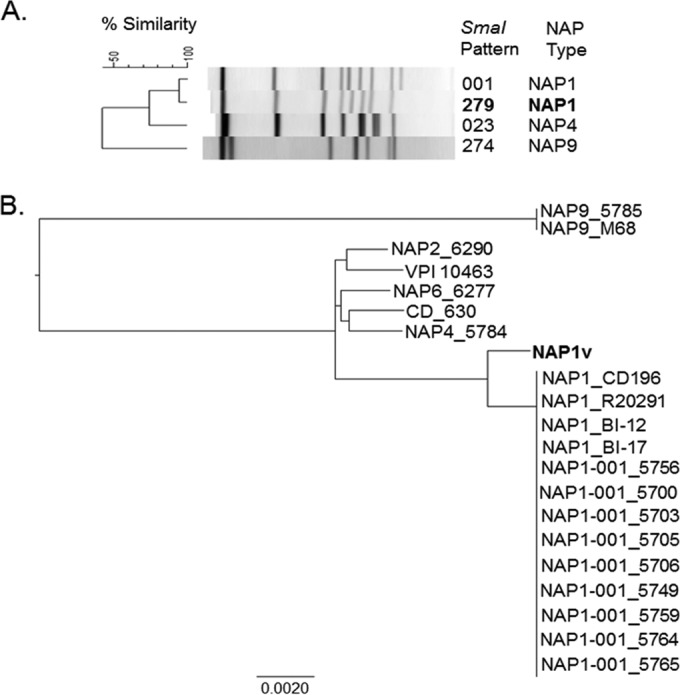
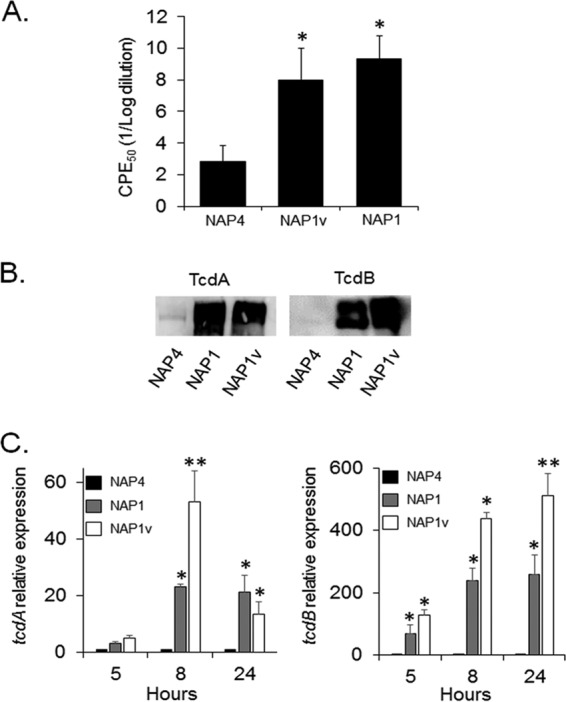
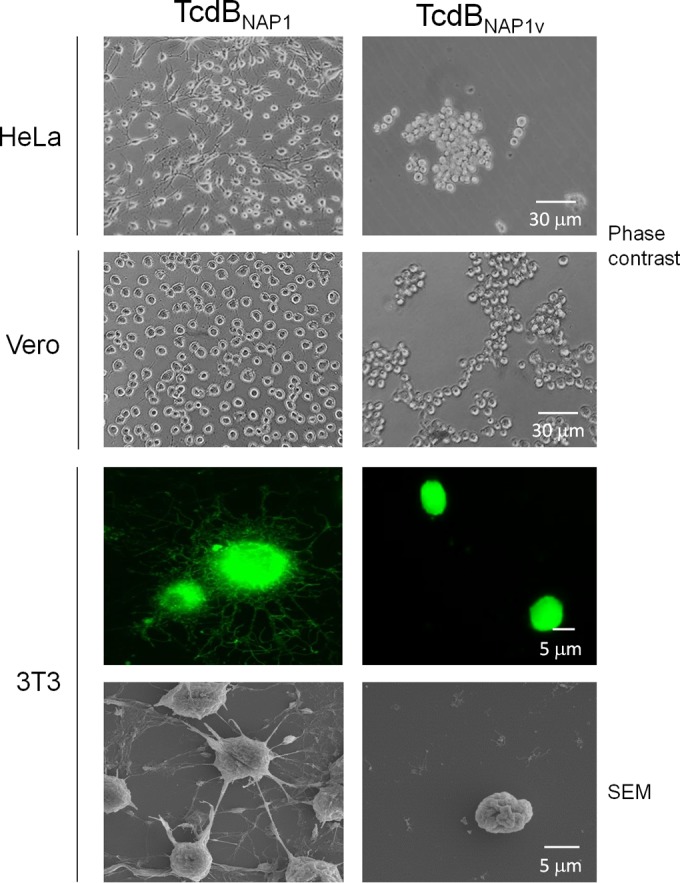
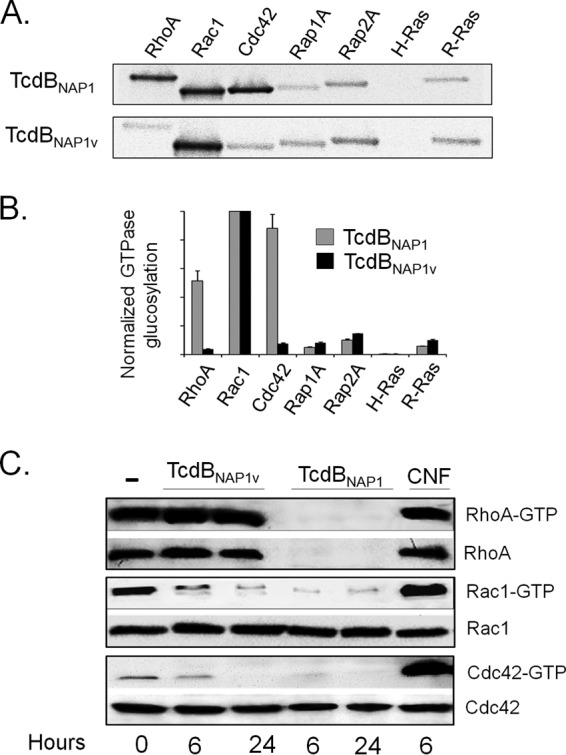
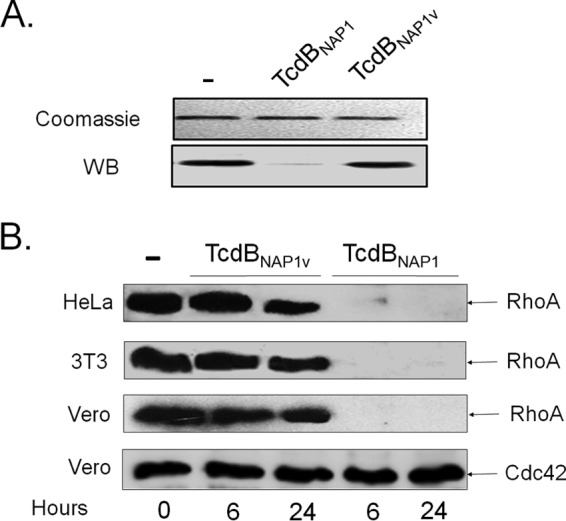
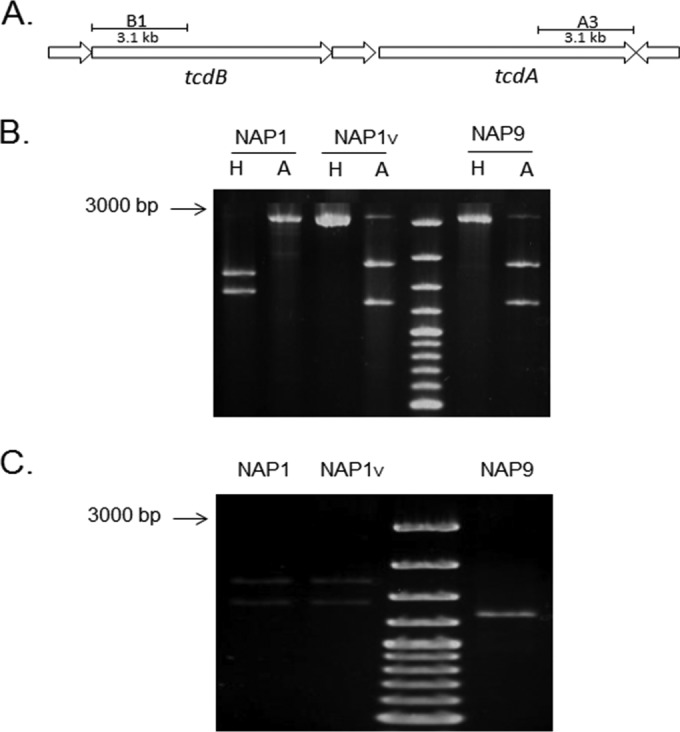
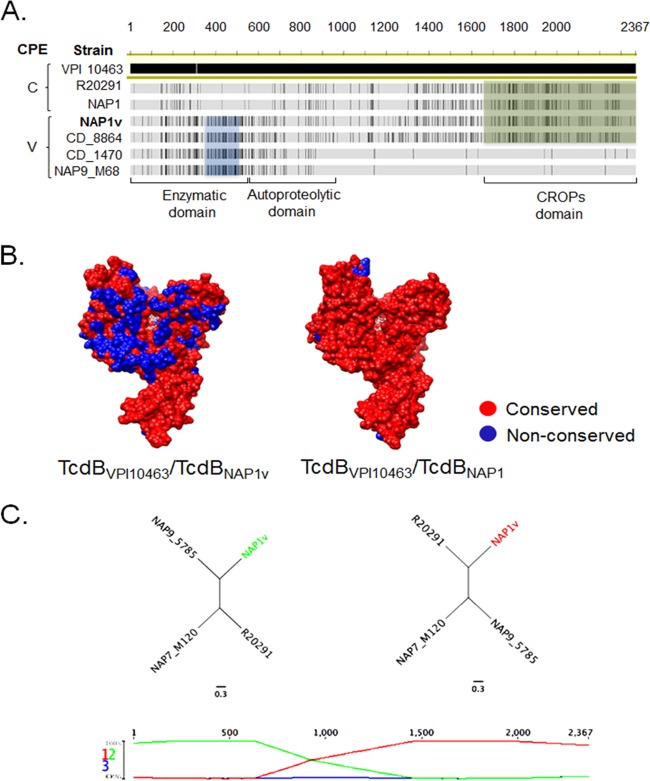
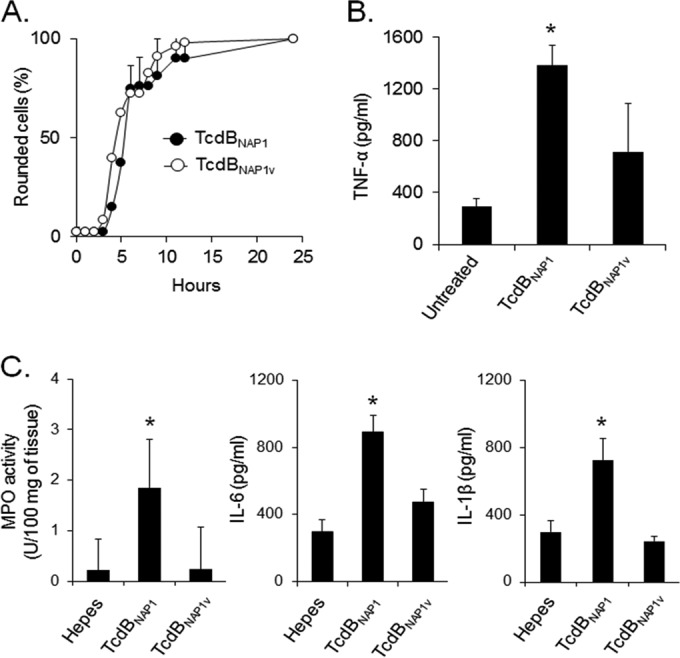
Clostridium difficile strains within the hypervirulent clade 2 are responsible for nosocomial outbreaks worldwide. The increased pathogenic potential of these strains has been attributed to several factors but is still poorly understood. During a C. difficile outbreak, a strain from this clade was found to induce a variant cytopathic effect (CPE), different from the canonical arborizing CPE. This strain (NAP1V) belongs to the NAP1 genotype but to a ribotype different from the epidemic NAP1/RT027 strain. NAP1V and NAP1 share some properties, including the overproduction of toxins, the binary toxin, and mutations in tcdC. NAP1V is not resistant to fluoroquinolones, however. A comparative analysis of TcdB proteins from NAP1/RT027 and NAP1V strains indicated that both target Rac, Cdc42, Rap, and R-Ras but only the former glucosylates RhoA. Thus, TcdB from hypervirulent clade 2 strains possesses an extended substrate profile, and RhoA is crucial for the type of CPE induced. Sequence comparison and structural modeling revealed that TcdBNAP1 and TcdBNAP1V share the receptor-binding and autoprocessing activities but vary in the glucosyltransferase domain, consistent with the different substrate profile. Whereas the two toxins displayed identical cytotoxic potencies, TcdBNAP1 induced a stronger proinflammatory response than TcdBNAP1V as determined in ex vivo experiments and animal models. Since immune activation at the level of intestinal mucosa is a hallmark of C. difficile-induced infections, we propose that the panel of substrates targeted by TcdB is a determining factor in the pathogenesis of this pathogen and in the differential virulence potential seen among C. difficile strains.
Copyright © 2016 Quesada-Gómez et al.
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
