

BayesFlow: latent modeling of flow cytometry cell populations
- PMID: 26755197
- PMCID: PMC4709953
- DOI: 10.1186/s12859-015-0862-z
BayesFlow: latent modeling of flow cytometry cell populations
Erratum in
-
Erratum to: BayesFlow: latent modeling of flow cytometry cell populations.BMC Bioinformatics. 2016 Mar 31;17:149. doi: 10.1186/s12859-016-0973-1. BMC Bioinformatics. 2016. PMID: 27036556 Free PMC article. No abstract available.
Abstract
Background: Flow cytometry is a widespread single-cell measurement technology with a multitude of clinical and research applications. Interpretation of flow cytometry data is hard; the instrumentation is delicate and can not render absolute measurements, hence samples can only be interpreted in relation to each other while at the same time comparisons are confounded by inter-sample variation. Despite this, most automated flow cytometry data analysis methods either treat samples individually or ignore the variation by for example pooling the data. A key requirement for models that include multiple samples is the ability to visualize and assess inferred variation, since what could be technical variation in one setting would be different phenotypes in another.
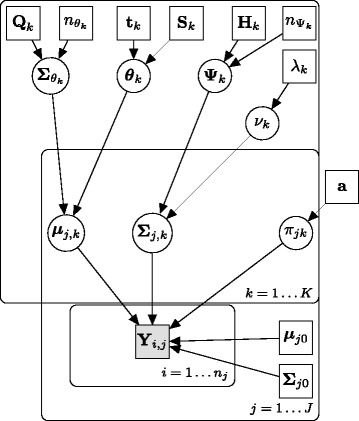
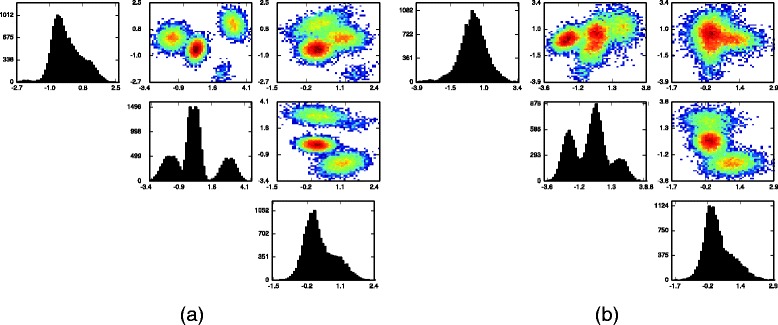
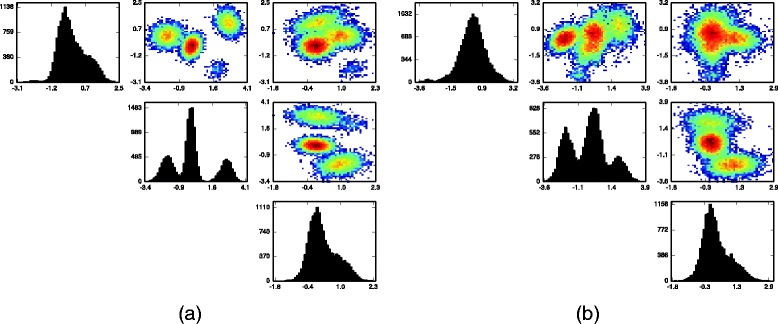
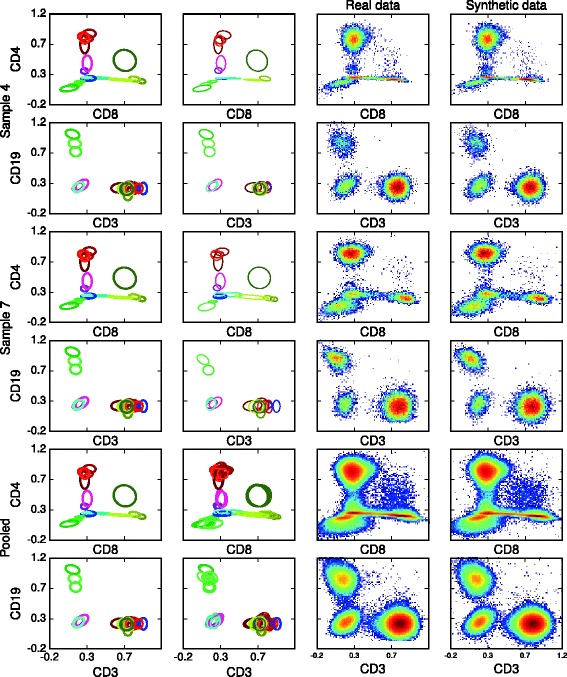
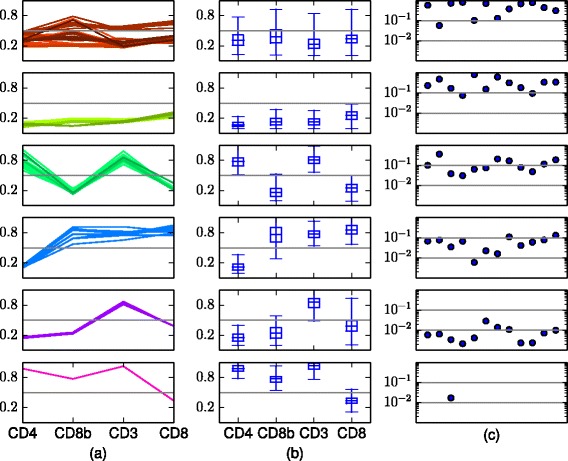
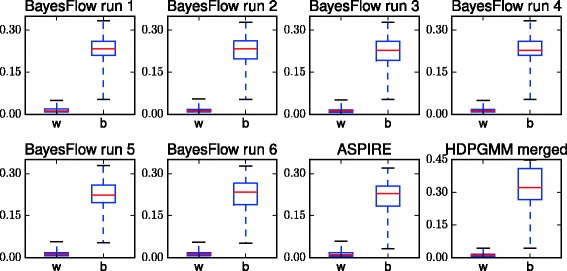
Results: We introduce BayesFlow, a pipeline for latent modeling of flow cytometry cell populations built upon a Bayesian hierarchical model. The model systematizes variation in location as well as shape. Expert knowledge can be incorporated through informative priors and the results can be supervised through compact and comprehensive visualizations. BayesFlow is applied to two synthetic and two real flow cytometry data sets. For the first real data set, taken from the FlowCAP I challenge, BayesFlow does not only give a gating which would place it among the top performers in FlowCAP I for this dataset, it also gives a more consistent treatment of different samples than either manual gating or other automated gating methods. The second real data set contains replicated flow cytometry measurements of samples from healthy individuals. BayesFlow gives here cell populations with clear expression patterns and small technical intra-donor variation as compared to biological inter-donor variation.
Conclusions: Modeling latent relations between samples through BayesFlow enables a systematic analysis of inter-sample variation. As opposed to other joint gating methods, effort is put at ensuring that the obtained partition of the data corresponds to actual cell populations, and the result is therefore directly biologically interpretable. BayesFlow is freely available at GitHub.
Figures
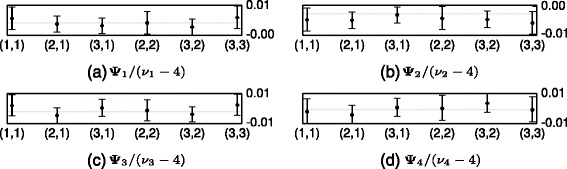
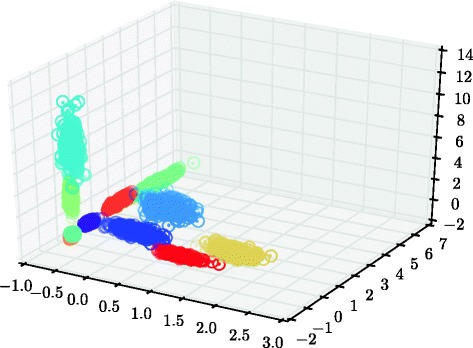
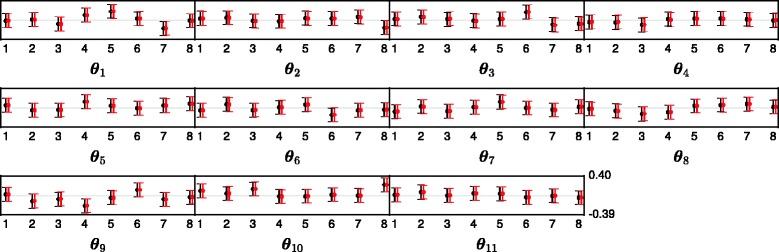
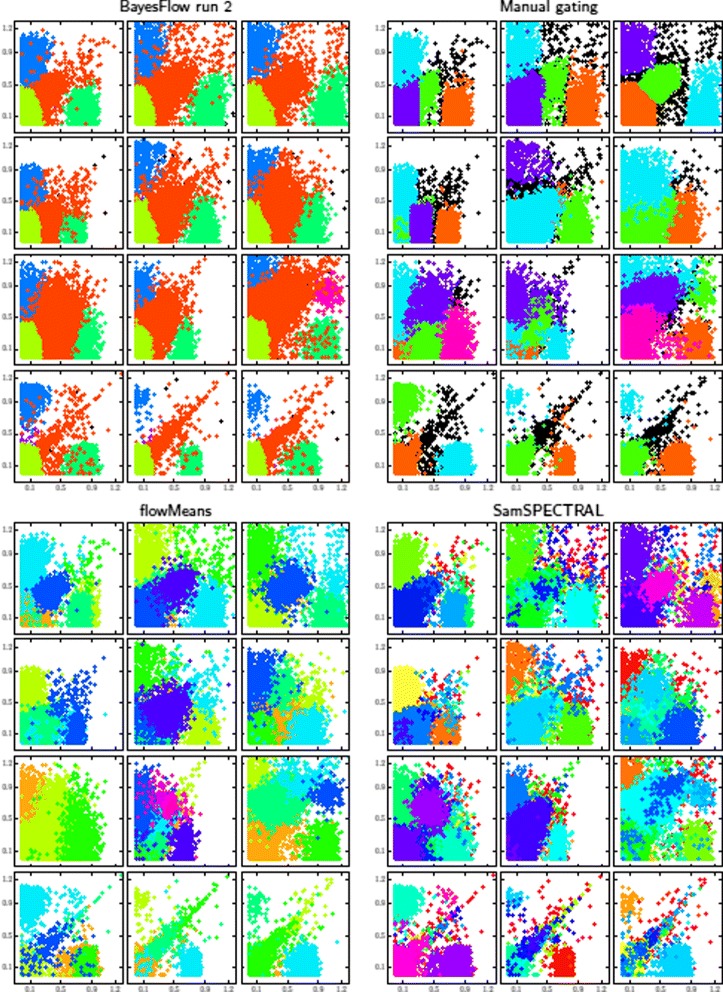
References
-
- Shapiro HM. Practical Flow Cytometry. Hoboken, New Jersey: John Wiley & Sons; 2005.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
