

Enhanced genome editing in mammalian cells with a modified dual-fluorescent surrogate system
- PMID: 26755436
- PMCID: PMC11108510
- DOI: 10.1007/s00018-015-2128-3
Enhanced genome editing in mammalian cells with a modified dual-fluorescent surrogate system
Abstract
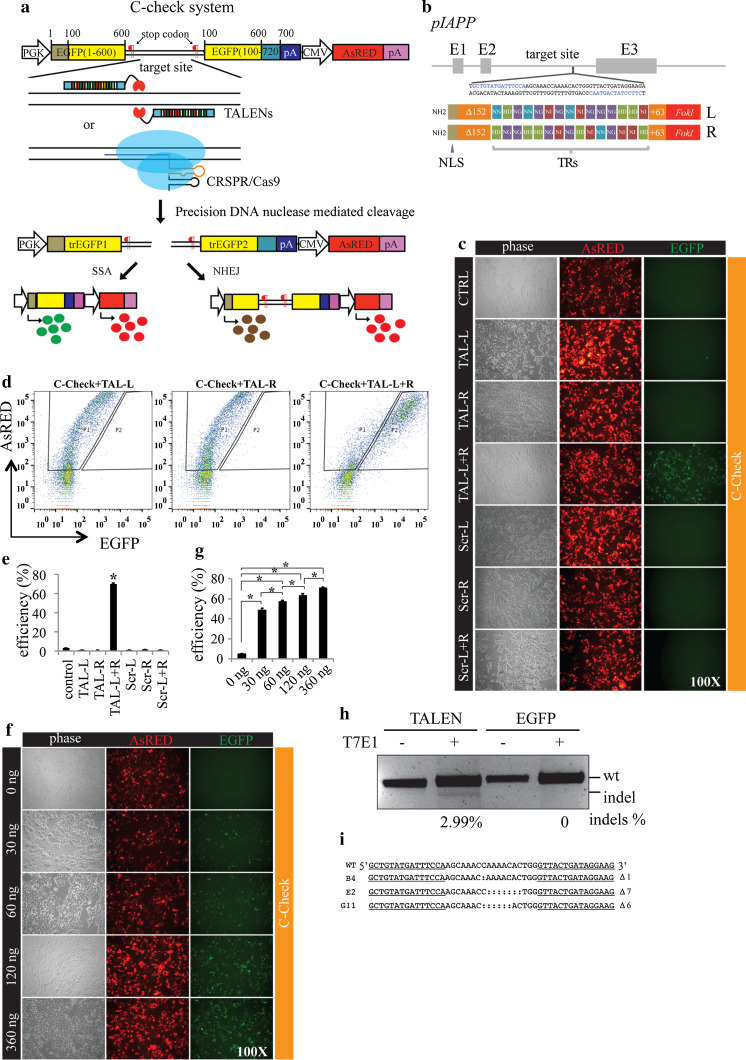
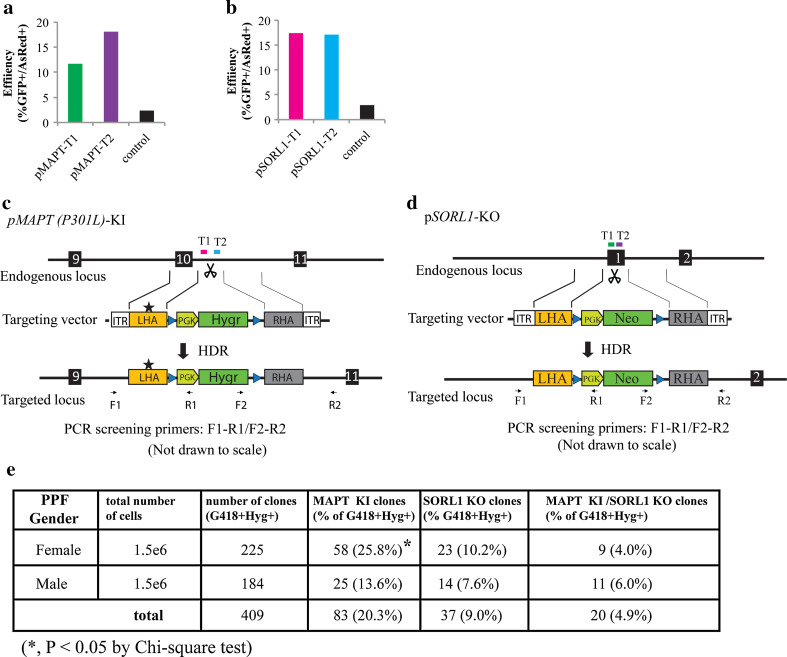
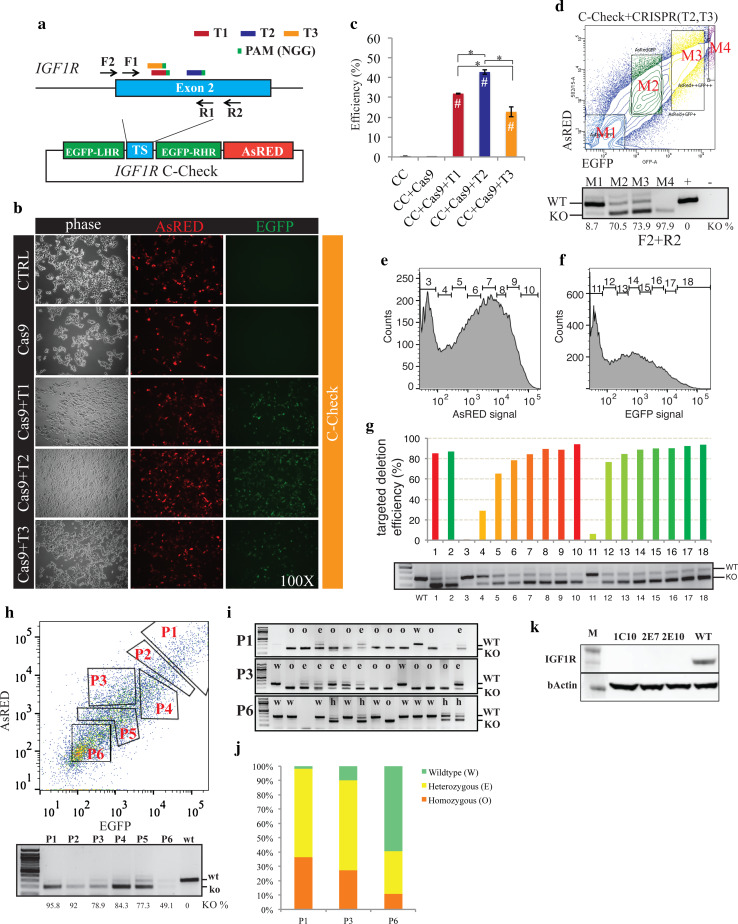
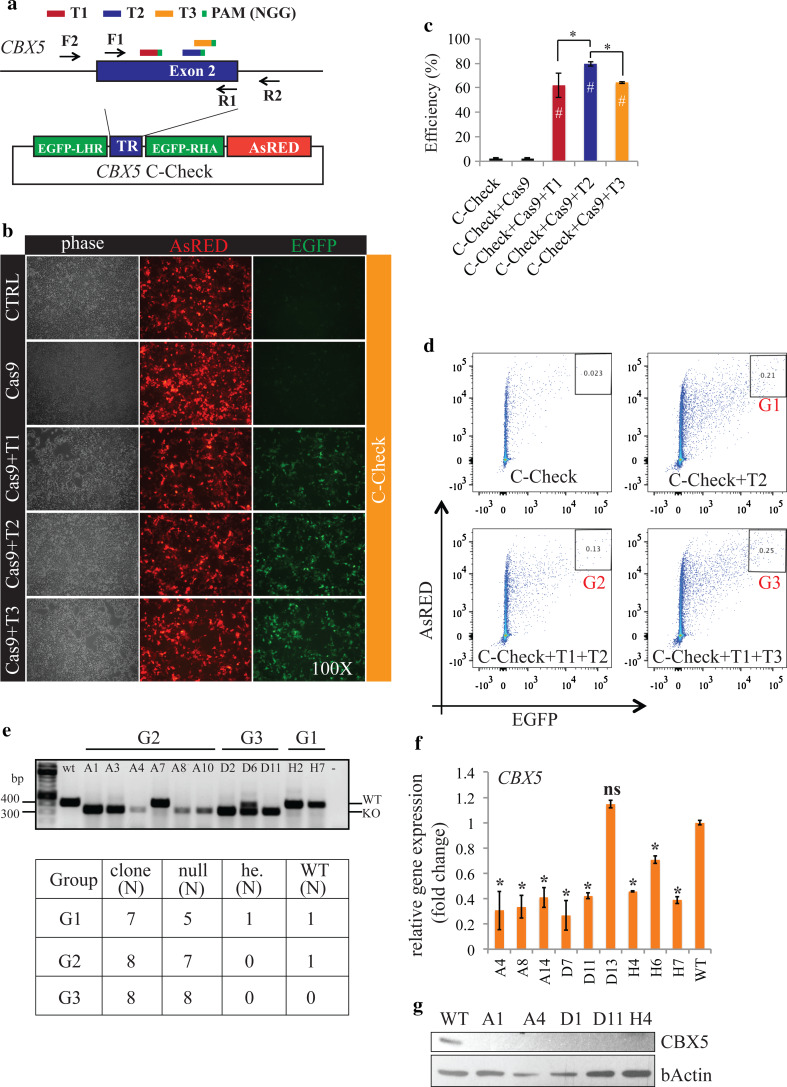
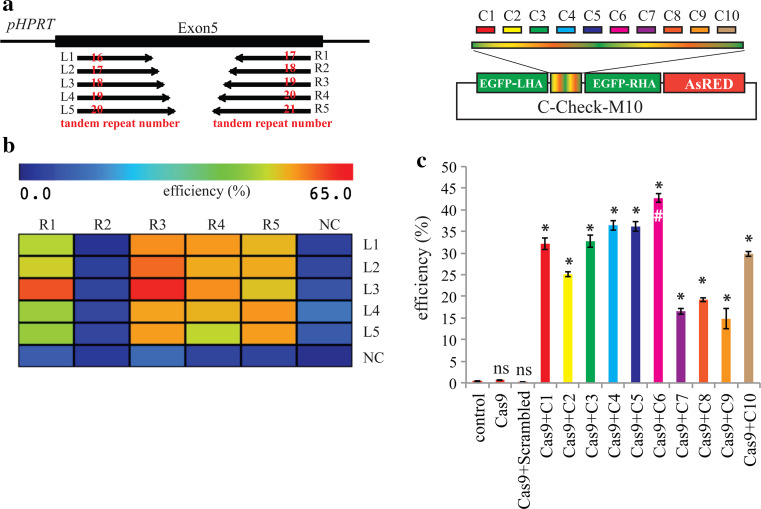
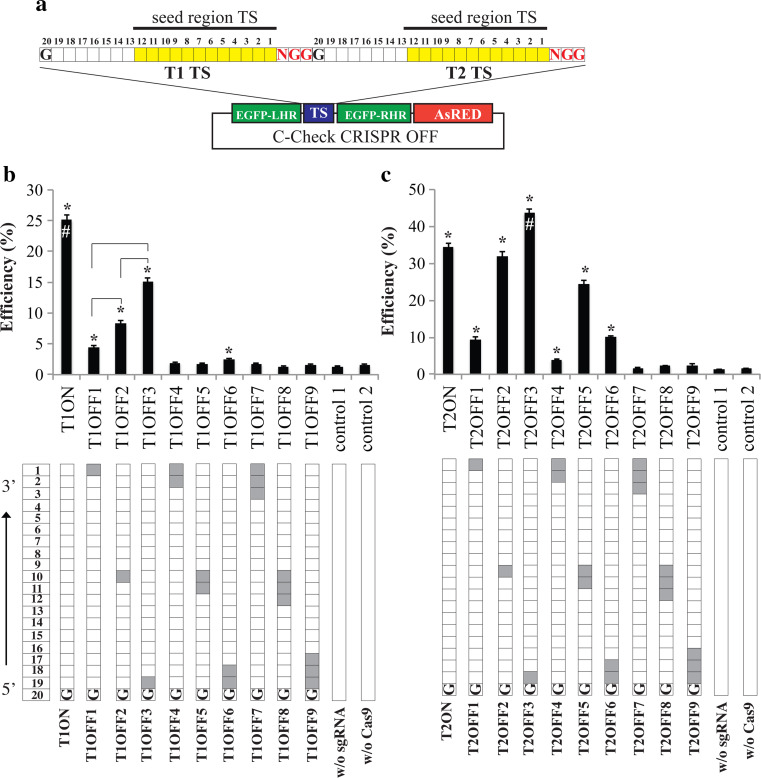
Programmable DNA nucleases such as TALENs and CRISPR/Cas9 are emerging as powerful tools for genome editing. Dual-fluorescent surrogate systems have been demonstrated by several studies to recapitulate DNA nuclease activity and enrich for genetically edited cells. In this study, we created a single-strand annealing-directed, dual-fluorescent surrogate reporter system, referred to as C-Check. We opted for the Golden Gate Cloning strategy to simplify C-Check construction. To demonstrate the utility of the C-Check system, we used the C-Check in combination with TALENs or CRISPR/Cas9 in different scenarios of gene editing experiments. First, we disrupted the endogenous pIAPP gene (3.0 % efficiency) by C-Check-validated TALENs in primary porcine fibroblasts (PPFs). Next, we achieved gene-editing efficiencies of 9.0-20.3 and 4.9 % when performing single- and double-gene targeting (MAPT and SORL1), respectively, in PPFs using C-Check-validated CRISPR/Cas9 vectors. Third, fluorescent tagging of endogenous genes (MYH6 and COL2A1, up to 10.0 % frequency) was achieved in human fibroblasts with C-Check-validated CRISPR/Cas9 vectors. We further demonstrated that the C-Check system could be applied to enrich for IGF1R null HEK293T cells and CBX5 null MCF-7 cells with frequencies of nearly 100.0 and 86.9 %, respectively. Most importantly, we further showed that the C-Check system is compatible with multiplexing and for studying CRISPR/Cas9 sgRNA specificity. The C-Check system may serve as an alternative dual-fluorescent surrogate tool for measuring DNA nuclease activity and enrichment of gene-edited cells, and may thereby aid in streamlining programmable DNA nuclease-mediated genome editing and biological research.
Keywords: CRISPR/Cas9; Dual-fluorescent surrogate reporter; Gene targeting; Genome engineering; Homologous recombination; Single-strand annealing; TALENs.
Conflict of interest statement
YL (2012–2014), SGR, and HD were financed by Novo Nordisk A/S. TK was financed by Gubra ApS. A patent claim is declared to the generation of a diabetes pig model based on genetic modification of the porcine
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
