

Mitochondrial Reactive Oxygen Species Mediate Cardiac Structural, Functional, and Mitochondrial Consequences of Diet-Induced Metabolic Heart Disease
- PMID: 26755553
- PMCID: PMC4859372
- DOI: 10.1161/JAHA.115.002555
Mitochondrial Reactive Oxygen Species Mediate Cardiac Structural, Functional, and Mitochondrial Consequences of Diet-Induced Metabolic Heart Disease
Abstract
Background: Mitochondrial reactive oxygen species (ROS) are associated with metabolic heart disease (MHD). However, the mechanism by which ROS cause MHD is unknown. We tested the hypothesis that mitochondrial ROS are a key mediator of MHD.
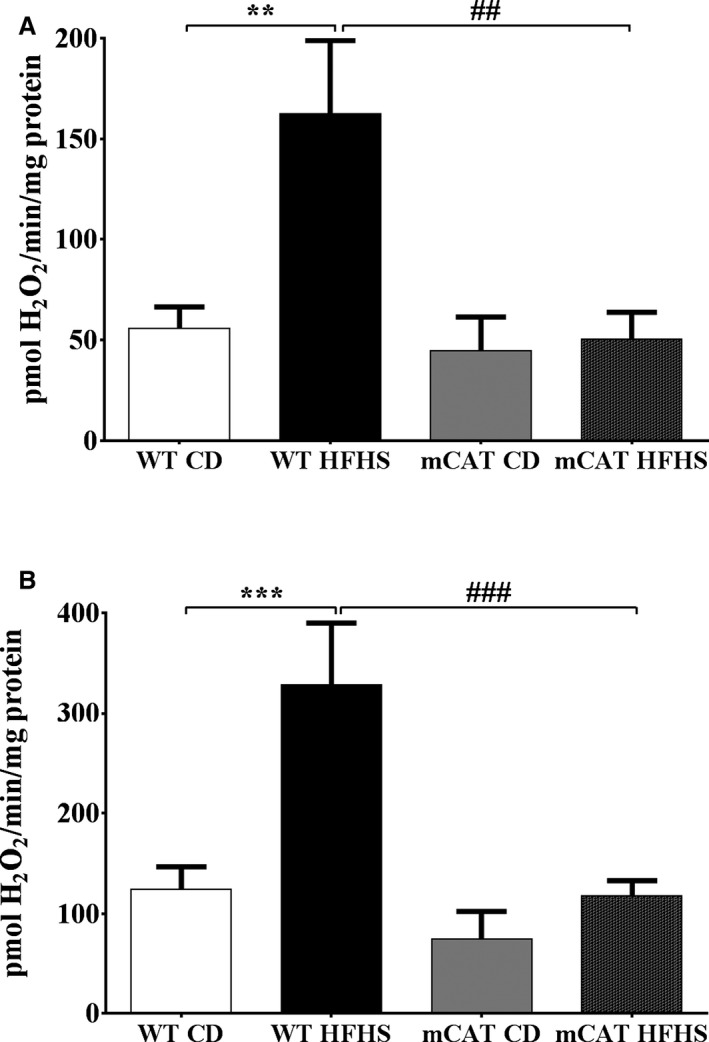
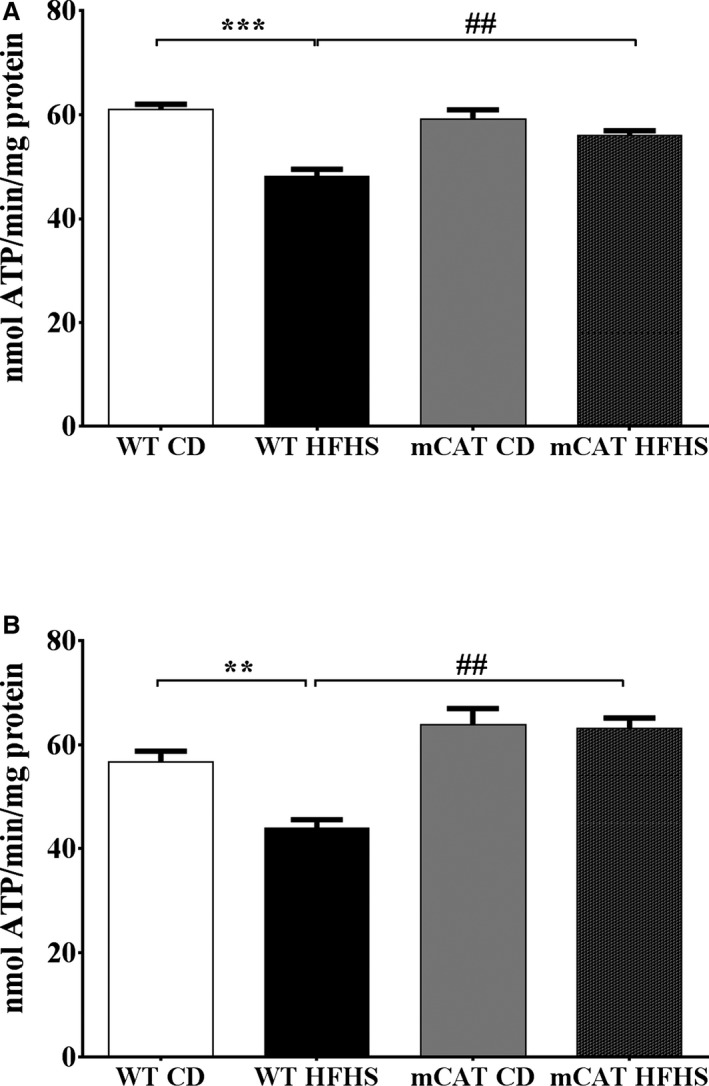
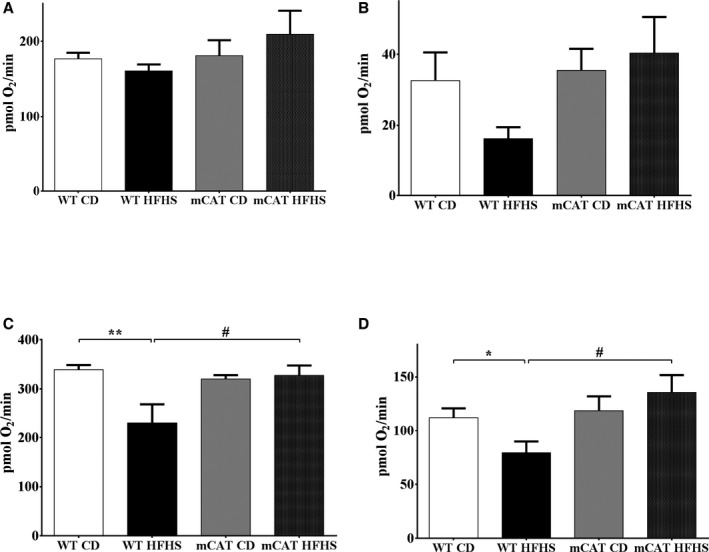
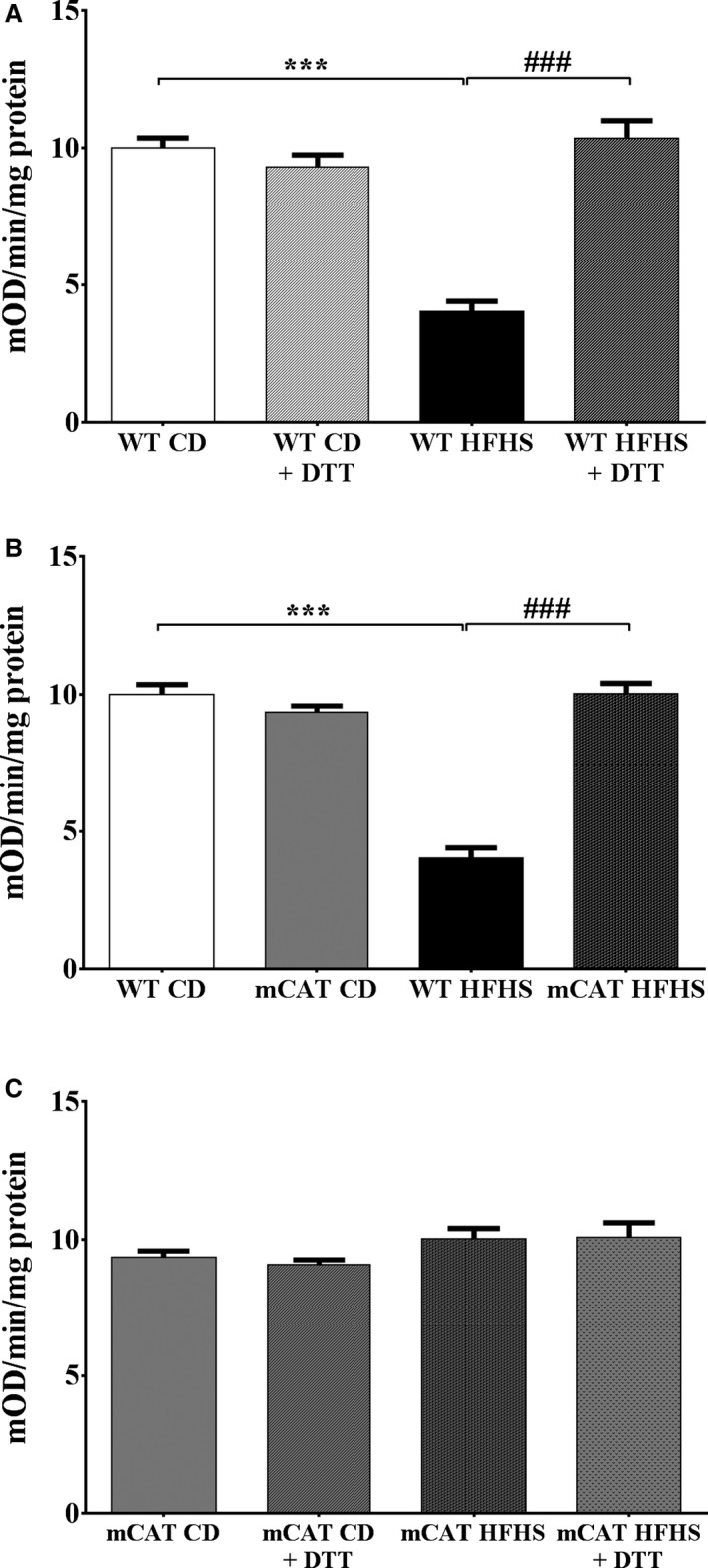
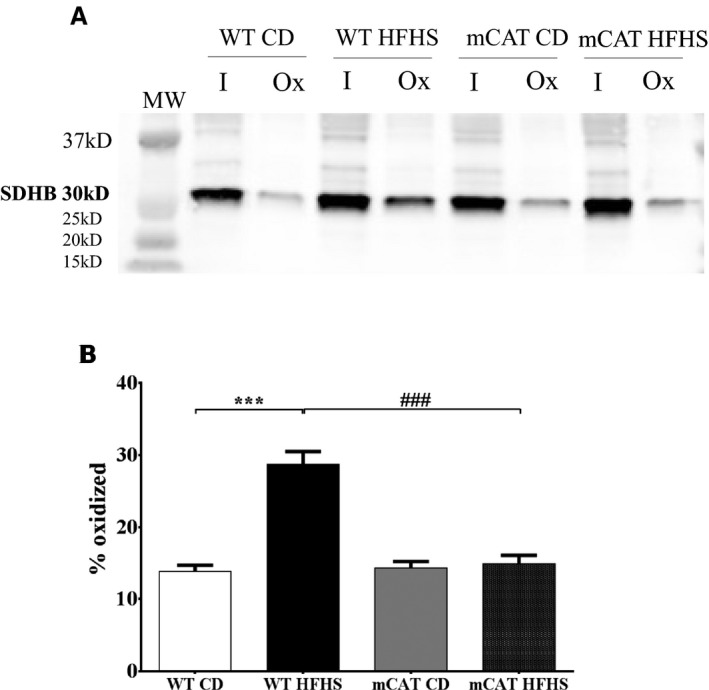
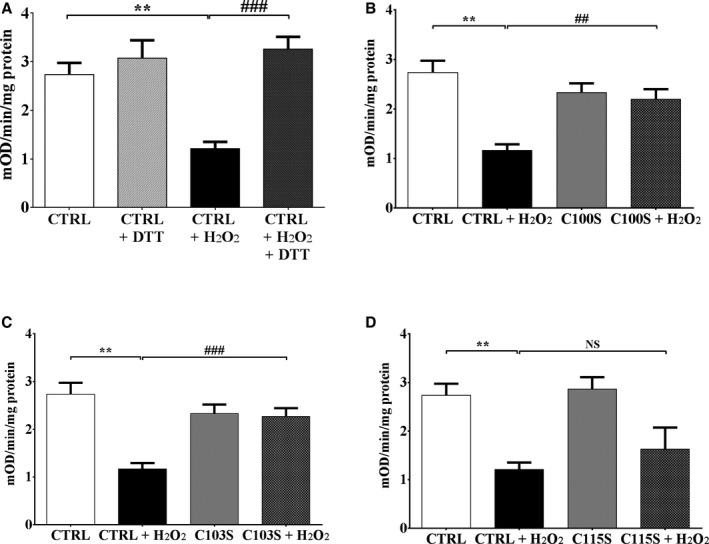
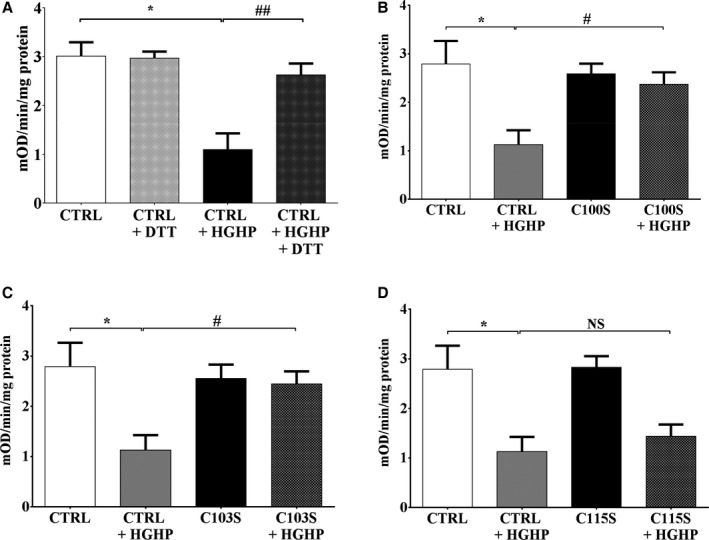
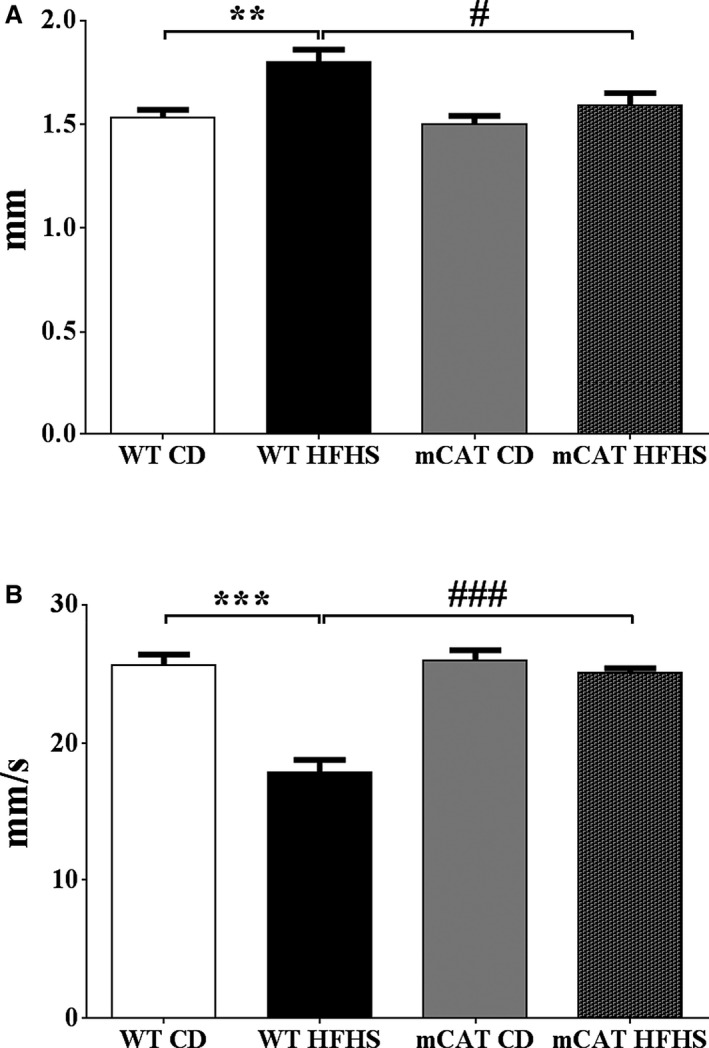
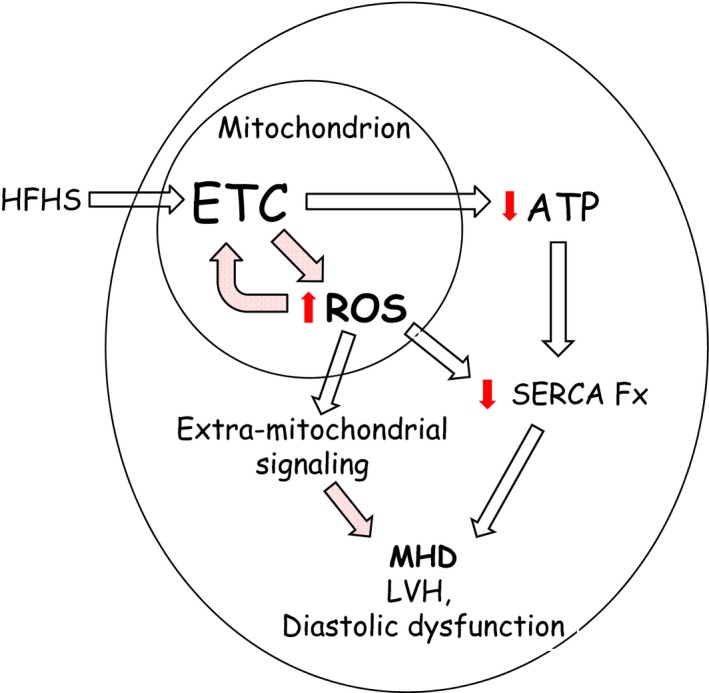
Methods and results: Mice fed a high-fat high-sucrose (HFHS) diet develop MHD with cardiac diastolic and mitochondrial dysfunction that is associated with oxidative posttranslational modifications of cardiac mitochondrial proteins. Transgenic mice that express catalase in mitochondria and wild-type mice were fed an HFHS or control diet for 4 months. Cardiac mitochondria from HFHS-fed wild-type mice had a 3-fold greater rate of H2O2 production (P=0.001 versus control diet fed), a 30% decrease in complex II substrate-driven oxygen consumption (P=0.006), 21% to 23% decreases in complex I and II substrate-driven ATP synthesis (P=0.01), and a 62% decrease in complex II activity (P=0.002). In transgenic mice that express catalase in mitochondria, all HFHS diet-induced mitochondrial abnormalities were ameliorated, as were left ventricular hypertrophy and diastolic dysfunction. In HFHS-fed wild-type mice complex II substrate-driven ATP synthesis and activity were restored ex vivo by dithiothreitol (5 mmol/L), suggesting a role for reversible cysteine oxidative posttranslational modifications. In vitro site-directed mutation of complex II subunit B Cys100 or Cys103 to redox-insensitive serines prevented complex II dysfunction induced by ROS or high glucose/high palmitate in the medium.
Conclusion: Mitochondrial ROS are pathogenic in MHD and contribute to mitochondrial dysfunction, at least in part, by causing oxidative posttranslational modifications of complex I and II proteins including reversible oxidative posttranslational modifications of complex II subunit B Cys100 and Cys103.
Keywords: metabolic heart disease; mitochondria; obesity; oxidative protein modifications; oxidative stress.
© 2016 The Authors. Published on behalf of the American Heart Association, Inc., by Wiley Blackwell.
Figures
References
-
- Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, Ikonomidis JS, Khavjou O, Konstam MA, Maddox TM, Nichol G, Pham M, Pina IL, Trogdon JG; American Heart Association Advocacy Coordinating C, Council on Arteriosclerosis T, Vascular B, Council on Cardiovascular R, Intervention, Council on Clinical C, Council on E, Prevention, Stroke C . Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail. 2013;6:606–619. - PMC - PubMed
-
- Main ML, Rao SC, O'Keefe JH. Trends in obesity and extreme obesity among US adults. JAMA. 2010;303:1695; author reply 1695‐1696. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
