

Absence of alsin function leads to corticospinal motor neuron vulnerability via novel disease mechanisms
- PMID: 26755825
- PMCID: PMC4764190
- DOI: 10.1093/hmg/ddv631
Absence of alsin function leads to corticospinal motor neuron vulnerability via novel disease mechanisms
Abstract
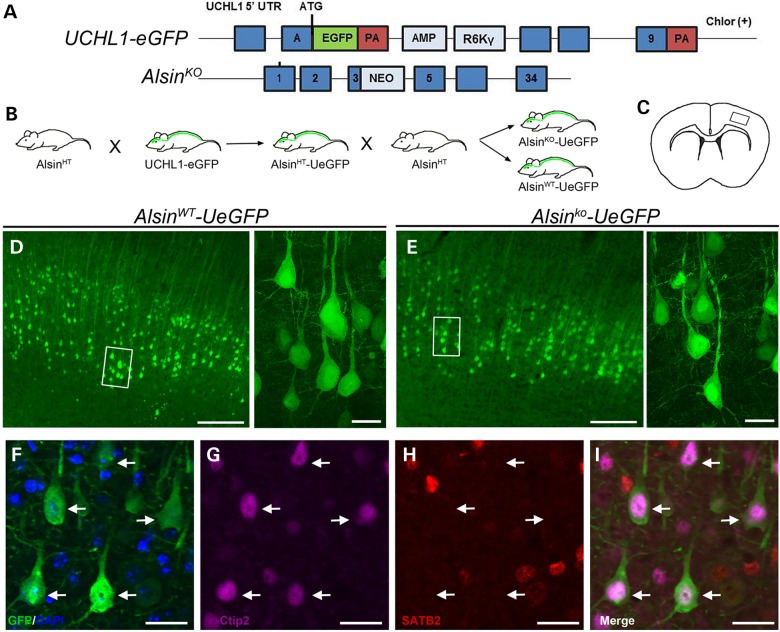
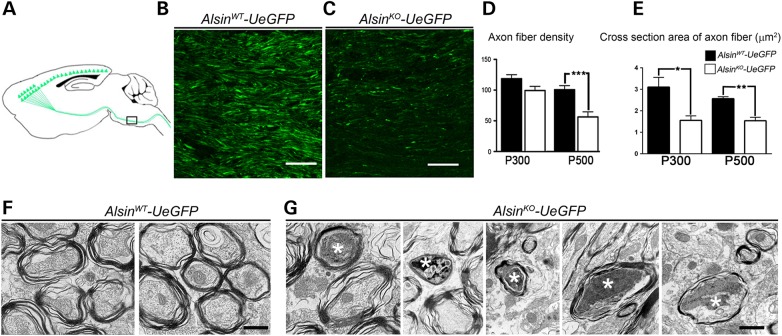
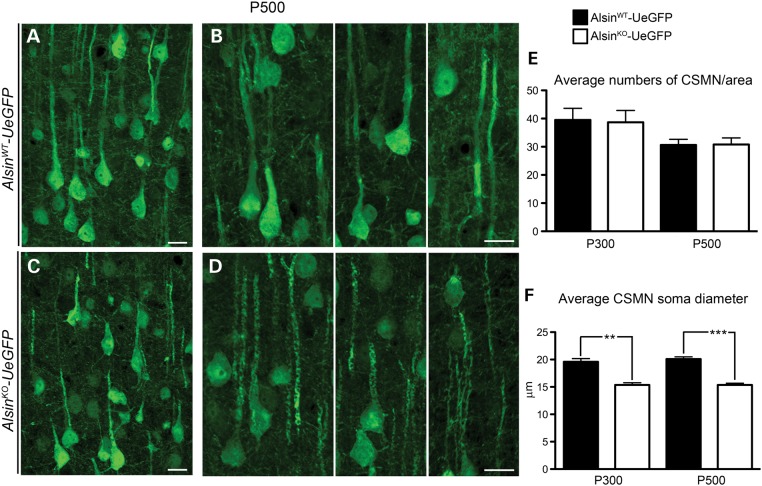
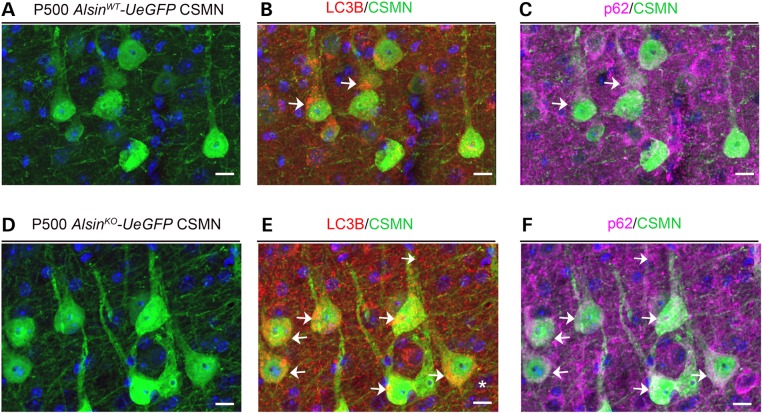
Mutations in the ALS2 gene result in early-onset amyotrophic lateral sclerosis, infantile-onset ascending hereditary spastic paraplegia and juvenile primary lateral sclerosis, suggesting prominent upper motor neuron involvement. However, the importance of alsin function for corticospinal motor neuron (CSMN) health and stability remains unknown. To date, four separate alsin knockout (Alsin(KO)) mouse models have been generated, and despite hopes of mimicking human pathology, none displayed profound motor function defects. This, however, does not rule out the possibility of neuronal defects within CSMN, which is not easy to detect in these mice. Detailed cellular analysis of CSMN has been hampered due to their limited numbers and the complex and heterogeneous structure of the cerebral cortex. In an effort to visualize CSMN in vivo and to investigate precise aspects of neuronal abnormalities in the absence of alsin function, we generated Alsin(KO)-UeGFP mice, by crossing Alsin(KO) and UCHL1-eGFP mice, a CSMN reporter line. We find that CSMN display vacuolated apical dendrites with increased autophagy, shrinkage of soma size and axonal pathology even in the pons region. Immunocytochemistry coupled with electron microscopy reveal that alsin is important for maintaining cellular cytoarchitecture and integrity of cellular organelles. In its absence, CSMN displays selective defects both in mitochondria and Golgi apparatus. UCHL1-eGFP mice help understand the underlying cellular factors that lead to CSMN vulnerability in diseases, and our findings reveal unique importance of alsin function for CSMN health and stability.
© The Author 2016. Published by Oxford University Press.
Figures

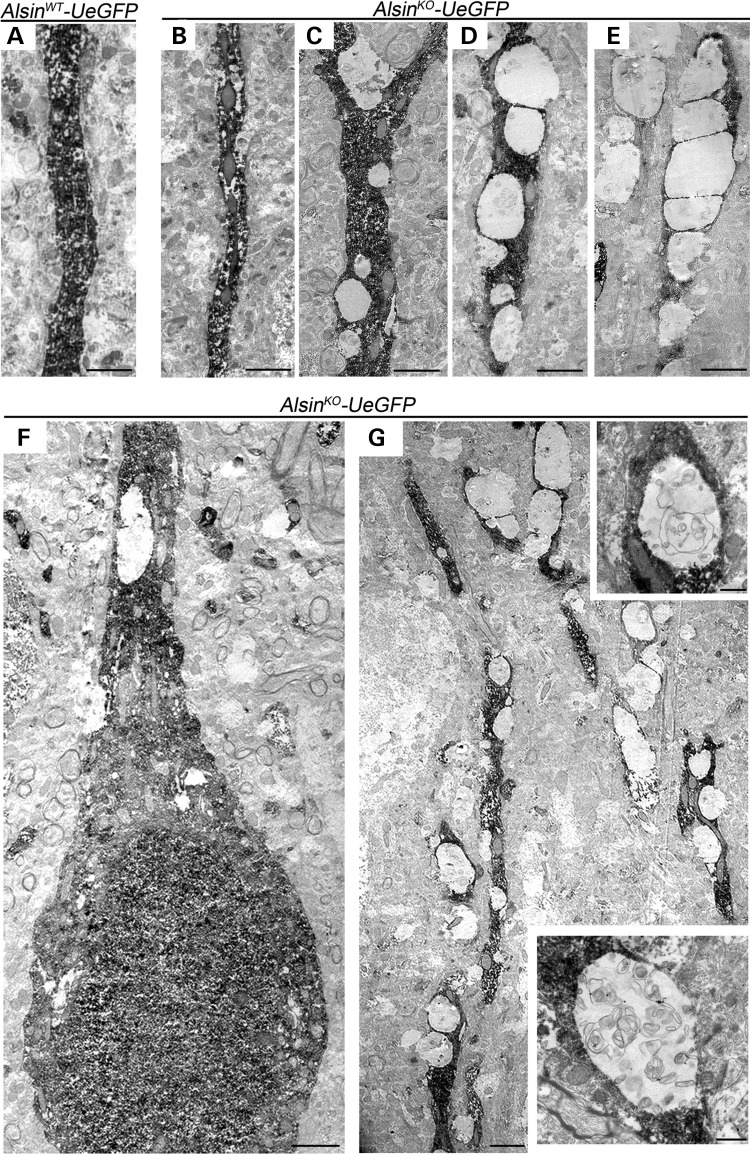
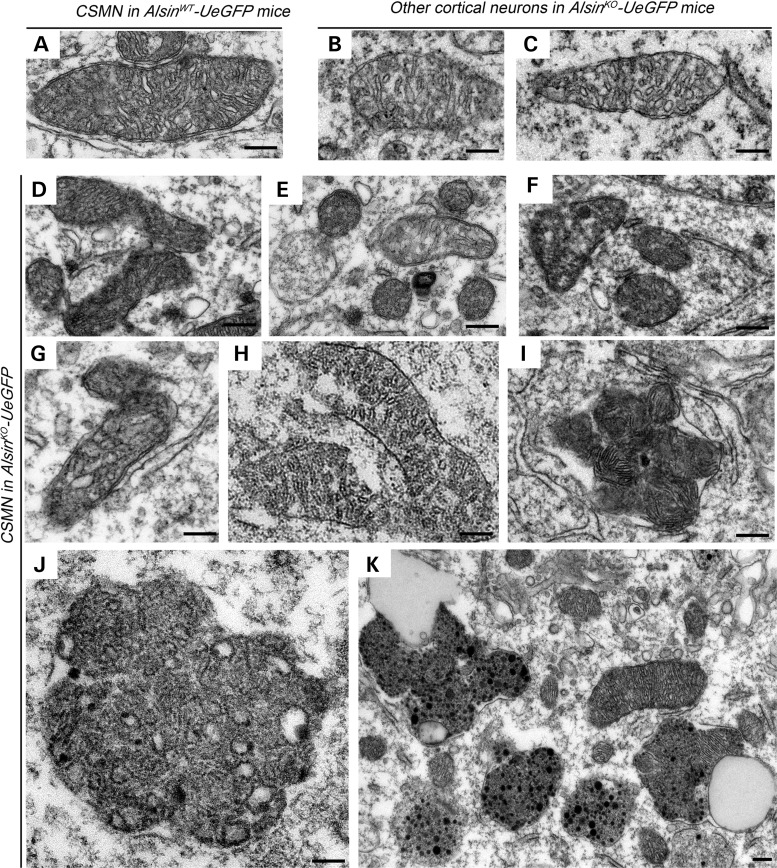
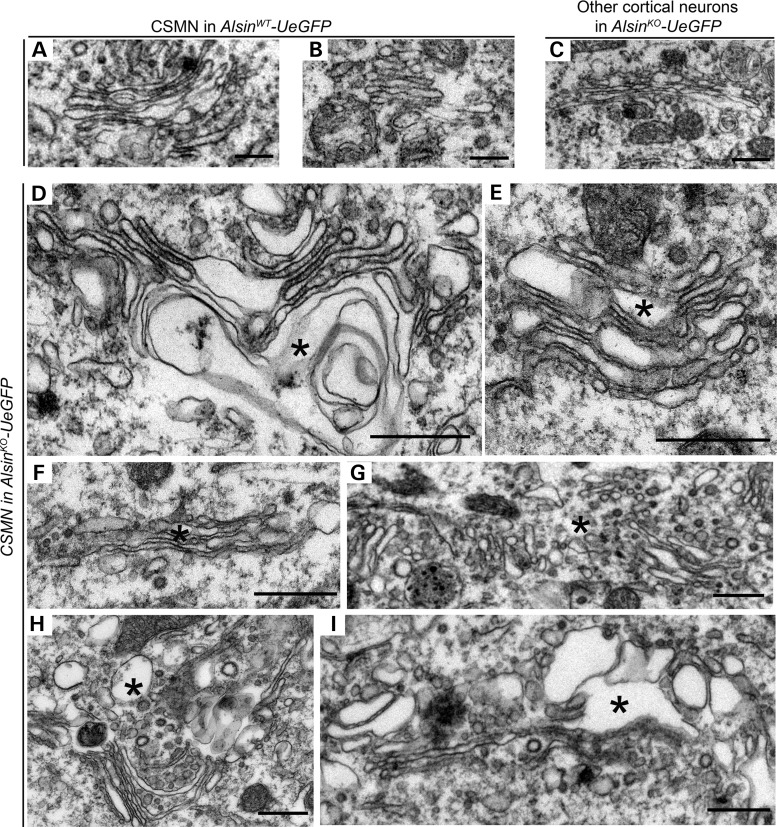
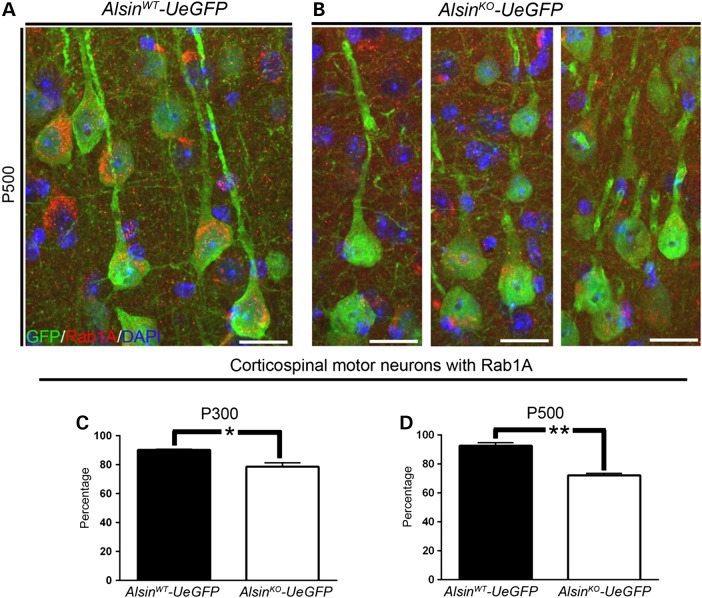
References
-
- Yang Y., Hentati A., Deng H.X., Dabbagh O., Sasaki T., Hirano M., Hung W.Y., Ouahchi K., Yan J., Azim A.C. et al. (2001) The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet., 29, 160–165. - PubMed
-
- Hadano S., Hand C.K., Osuga H., Yanagisawa Y., Otomo A., Devon R.S., Miyamoto N., Showguchi-Miyata J., Okada Y., Singaraja R. et al. (2001) A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet., 29, 166–173. - PubMed
-
- Eker H.K., Unlu S.E., Al-Salmi F., Crosby A.H. (2014) A novel homozygous mutation in ALS2 gene in four siblings with infantile-onset ascending hereditary spastic paralysis. Eur. J. Med. Genet., 57, 275–278. - PubMed
-
- Wakil S.M., Ramzan K., Abuthuraya R., Hagos S., Al-Dossari H., Al-Omar R., Murad H., Chedrawi A., Al-Hassnan Z.N., Finsterer J. et al. (2014) Infantile-onset ascending hereditary spastic paraplegia with bulbar involvement due to the novel ALS2 mutation c.2761C>T. Gene, 536, 217–220. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
